{kind=link}

Nature. Vol. 345. 7 June, 1990
ARTICLES
Click Here to See Cover Photo
David Vaux*, John Tooze & Stephen
Fuller ^ #
* Cell Biology and ^ Biological Structures Programmes,
European Molecular Biology Laboratory,
Meyerhofstrasse 1, 6900 Heidelberg, FRG
# To whom correspondence should be addressed.
Monoclonal antibodies were raised against antibodies to distinct carboxy terminal KDEL sequences of two soluble, resident endoplasmic reticulum proteins. These anti-idiotype reagents recognize an intrinsic membrane protein with characteristics expected of a receptor responsible for the recognition and return of resident proteins to the endoplasmic reticulum.
EUKARYOTIC secretory proteins begin their journey out of the cell by co-translational translocation into the lumen of the endoplasmic reticulum (ER)1. Vesicular transport 2 of these soluble proteins to the Golgi apparatus seems to be by a nonspecific fluid phase transport that does not require a specific signal 3. The presence of several resident soluble proteins in the lumen of the ER provides an apparent exception to this hypothesis of nonspecific fluid phase transport 4. Several of these resident proteins mediate the proper assembly and folding of newly synthesized proteins. They include the immunoglobulin heavy chain-binding protein (BiP), which apparently functions in protein oligomerization 5, and protein disulphide isomerase (PDI), which catalyses the rearrangement of disulphide bonds 6. These retained soluble ER proteins are not a minor population; from data in the literature 7,8 we calculate that PDI is a very abundant protein of the ER, present at a concentration of >400 µM.
Peptides were synthesized in an automated continuous flow instrument using FMOC chemistry 29 and purified by reverse phase HPLC. The peptide KXXXXXKDEL consists of a mixture of analogues containing any one of alanine, aspartic acid, histidine, glutamine, leucine, tyrosine or lysine at each of the positions X. KDEL-terminated peptides are identified throughout by their unique amino-terminal residues. Single-letter amino-acid code used.
A clue to the mechanism of retention in the ER came from the finding that BiP and PDI share a carboxy-terminal sequence (Table 1), Lys-Asp-Glu-Leu (KDEL) with other resident ER proteins including glucose-regulated protein 94 (GRP 94) (ref. 9). In addition, a secreted soluble protein, lysozyme, can be converted into a retained protein of the ER by adding the last six amino acids of BiP, Ser-Glu-Lys-Asp-Glu-Leu, to the carboxy terminus. Addition of a further two amino acids (GlyLeu; GL) to the carboxy terminus results in secretion of the mutant Iysozyme, as do numerous other changes in the KDEL terminus 10. Thus, the presence of KDEL at the carboxy terminus is sufficient for the retention of a protein in the ER. A similar retention system functions, albeit less efficiently, in the yeast Saccharomyces cerevisiae 11, although a different recognition signal, His-Asp-Glu-Leu (HDEL), is used.
The retention of resident proteins bearing the KDEL signal by interaction with a static receptor within the ER seems unlikely: the proteins that must be retained by this receptor are far more abundant than any single candidate receptor polypeptide. It has been suggested that the receptor might function in a distal compartment by recognizing the KDEL signal on escaping resident proteins and mediating their return to the ER 10. Microinjection experiments 12 demonstrate that the presence of a KDEL tail on BiP does not significantly alter its rate of diffusion in the ER from that of BiP bearing the unrecognized KDELGL tail. Both diffuse more rapidly than the integral membrane protein, haemagglutinin, but more slowly than the secreted protein, albumin. Hence, the KDEL signal is not continually and tightly bound to a component of the ER membrane although proteins bearing this signal probably interact weakly within the ER. Proteins with the KDEL signal do leave the ER transiently, as shown by the modification of a cathepsin D hybrid protein bearing the KDEL tail 13. Thus, resident ER proteins presumably pass through a distal compartment that is proximal to the classical Golgi apparatus before returning to the ER 4. Compartments have been identified between the ER and classical Golgi by a variety of approaches 14-16. Although the relationship between these structures remains unclear, we will refer to them collectively as the salvage compartment 4.
The identification of a receptor for the KDEL signal must rest on several assumptions about its properties. First, we assumed that all KDEL signals must be recognized by a single receptor species that does not bind unrecognized sequences such as K . . . GL. Second, we assumed that the receptor is a recycling protein and must have two distinct binding states. While functioning, the receptor must alternate between a high-affinity state that recognizes escaped KDEL-bearing proteins and a low affinity state that releases them into the lumen of the ER. Conventional ligand affinity methods for receptor identification are problematic because the ionic environment of the ER and salvage compartment are not well defined. We made broad assumptions about receptor localization. The receptor could be present in the ER, in transitional elements, in the salvage compartment, in proximal parts of the Golgi apparatus and in transport vesicles between these compartments. For these reasons we used an immunological approach to search for a protein with the properties required of the receptor.
Monoclonal internal image anti-idiotype antibodies are powerful tools for the identification and characterization of the participants in a receptor-ligand interaction 17-18. Peptides containing the KDEL signal (Table 1) can be used to raise anti-KDEL antibodies that recognize this signal at the carboxy terminus of ligands. Internal image anti-idiotype antibodies 18-20 raised against these anti-KDEL antibodies should then recognize the receptor by its KDEL-binding site. We raised such anti-idiotype antibodies and demonstrate here that they recognize an intrinsic membrane protein that is a candidate for the KDEL receptor.
Antibody generation
Synthetic peptides corresponding to a variety of carboxy-terminal
sequences (Table 1) were used to generate polyclonal and monoclonal
antibodies. Rabbit antisera to some of these peptides have already
been shown to recognize the corresponding ER proteins and to label
the ER by electron microscope (EM) immunocytochemistry 21;
a more complete characterization of these antibodies will be presented
elsewhere (S.F., J.T. and D.V., manuscript in preparation). An
important assumption of the anti-idiotype approach is that the
first round antibodies recognize the KDEL retention signal itself
on resident ER proteins. As the signal specificity lies within
the last four residues, recognition by our first round antibodies
must be dependent on the carboxy terminus. A monoclonal anti-PDI
tail antibody was generated by immunization with peptide KDDD
(Table 1). This IgG antibody (1D3) recognizes a double band of
relative molecular mass ~55,000 (Mr~ 55K) in western
blots, consisting of PDI and calreticulin 22, labels
the ER by EM immunocytochemistry 21 and gives a clear
reticular pattern by immunofluorescence (Fig. 7h). Figure 1 confirms
that the signal recognized by 1D3 can be removed from PDI by gentle
incubation with carboxypeptidase A under conditions that do not
change the apparent protein pattern of the blotted lysate and
do not affect the recognition of PDI by a polyclonal antiserum
raised against rat PDI. Thus, 1D3 recognition depends on carboxy-terminal
residues. Taken in conjunction with solid phase assays on a range
of synthetic peptides (data not shown), this result leads us to
conclude that 1D3 recognizes the retention signal in PDI and calreticulin.
Similarly, polyclonal antisera to the KETE peptide (Table I) recognize
specifically the carboxy-terminal residues of BiP, PDI, GRP 94
and other proteins of the ER (S.F., J.T. and D.V., manuscript
in preparation).
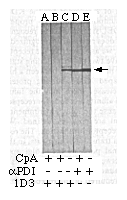
FIG. 1 Monoclonal antibody, 1D3, recognizes
a carboxy-terminal determinant on an ER protein, PDI. Western
blot using monoclonal antibody 1D3 (lanes A, B, C) or rabbit anti-PDI
antiserum (lanes D, E) on total cellular protein before (lanes
C, E) and after treatment with carboxypeptidase A at low concentration
(lane A) or at high concentration (lanes B, D). The arrow identifies
PDI.
METHODS. VG mouse hybridoma total cell protein was separated by
gel electrophoresis30 and electrophoretically transferred
to nitrocellulose using a Genie electroblotter (Idea Scientific,
Inc.) according to the manufacturers instructions. Strips from
a single blot were washed in 2% (v/v) Tween20 in 25 mM Tris-HCl
buffer 150 mM NaCl (TN), pH 7.6, and incubated overnight at 37°C
in 25 ml of 2% Tween-20 TN pH 5.5 buffer, other incubations contained
a 1:250 or a 1:2500 dilution of carboxypeptidase A (Sigma). The
filters were then washed in 2% Tween-20 TN pH 7.6, blocked with
BLOTTO31, and probed using either monoclonal antibody
1D3 (prepared against KDDD peptide coupled to Keyhole Limpet Hemocyanin
(KLH)) or a rabbit antiserum to PDI (rabbit immunized as described
21 with protein purified by the method of Lambert and
Freedman 32) followed by appropriate affinity purified
alkaline phosphatase-conjugated goat second antibodies 33.
Two-dimensional gel electrophoresis followed by western blotting
with anti-KEEE antiserum confirmed that the second band recognized
by 1D3 is calreticulin (lane C).
Two approaches were used to prepare anti-idiotype antibodies to the anti-tail antibodies. First, monoclonal anti-idiotype antibodies were generated by the method of paired in vitro immunization 18 using unconjugated KETE peptide as the starting immunogen. The second-round hybridomas were screened for ER- or Golgi-related staining patterns by indirect immunofluorescence. Two wells were selected for further characterization, both of which yielded stable IgM-secreting hybridoma cell lines after cloning in agarose. The results obtained with the line designated 5D3 are described below. Second, syngeneic mice were immunized with fixed cells of the 1D3 hybridoma cell line. Hybridomas resulting from this in vivo immunization were screened for reactivity with anti-KDEL antibodies (rabbit anti-KAVK). Of 256 hybrid-containing wells, 15 gave a strong reaction. The results for a stable, cloned IgG-secreting line from one of these, 9D6, are described below.
Anti-anti-KDEL antibodies
Immunoblotting of lysates of control murine hybridoma cells
(VG cells) revealed a major immunoreactive band of 72K with 5D3
and 9D6 (Fig. 2a). This 72K antigen remained with the membrane
pellet after carbonate-washing of microsomes up to pH 11 (Fig.
2e). The 72K antigen also partitioned into the detergent phase
on separation of a TX-114 lysate of VG cells (not shown). Thus,
the antigen is presumably an integral membrane protein 23.
Protease protection experiments revealed that a 54K immunoreactive
fragment remained resistant to protease treatment of intact VG
cell microsomes (Fig. 2c). Neither band was present when protease
treatment followed detergent lysis, and blotting with anti-PDI
showed that this luminal ER protein was completely protected under
the conditions of Fig. 2c, lane 2 (not shown). Immunoblotting
with 5D3 showed that the antigen was present in all the vertebrate
species tested but not in S. cerevisiae (data not shown).
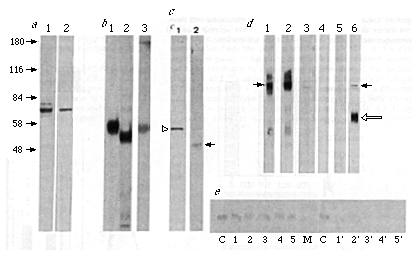
FIG. 2 Both monoclonal anti-idiotype antibodies
recognize the same integral membrane protein. The total post-nuclear
lysate from the mouse hybridoma cell line VG was blotted with
the monoclonal antibodies 5D3 (a, lane 1) and 9D6 (a,
lane 2). Mr (in K) marker positions for a are
indicated to the left. Antigen was immuno-precipitated from [35S]methionine-labelled
9D6 (b, lane 1) or 5D3 (b, lane 2) cell lysates. The cross
reactivity of the 5D3 and 9D6 antigens is demonstrated by the
isolation of the immunoglobulin-antigen complex from supernatant
of 9D6 hybridomas with fixed S. aureus cells and immunoblotting
with 5D3 (b, lane 3). VG microsomes treated with proteinase K
and blotted with 5D3 show that a cytoplasmic portion of the antigen
is accessible to cleavage and that the antigenic site is contained
within the protected lumenal domain (c, lane 2, solid arrow;
undigested antigen, c, lane 1 open arrow). Blotting of
an equal amount of protein from total post-nuclear lysates from
5D3 (d, lane 1), 9D6 (d, lane 2) or VG (d,
lane 3) with the monoclonal antibody 5D3 shows the increased level
of antigen (solid arrows) expression in the anti-idiotype hybridomas.
VG lysate was immunoblotted with 1D3 ascites using a µ-chain
specific antibody to show IgM reactivity (d, lane 5), which
was absent when 1D3 supernatant was used (d, lane 4). VG
lysate was also western blotted with polyclonal mouse antisera
raised against the isolated 5D3 antigen (d lane 6). The position
of the mouse heavy chain in VG cell lysates is marked by the open
arrow. e, Demonstration of the membrane-associated nature
of the 5D3/9D6 antigen by carbonate washing of VG microsomes at
various pH (lane 1, pH 11.6; 2, pH 11.0; 3, pH 10.5; 4, pH 10.0;
5, pH 9.5) followed by western blotting of the pellet (lanes 1-5)
or supernatant (lanes 1'-5') with 5D3. The 84K marker (lane M)
and noncarbonate-treated microsomes (lane C) are also shown.
METHODS. Monoclonal antibody 5D3 was produced by paired in vitro
immunization 18 using 100 µg unconjugated HPLC-purified
KETE peptide as starting antigen. Monoclonal antibody 9D6 was
produced by fusing Ag8 myeloma cells to spleen cells from a BALB/c
mouse immunized with washed 3% (w/v) paraformaldehyde-fixed 1D3
(anti-PDI tail) hybridoma cells 34. Washed VG cells
at 10 7 ml -1 were lysed in lysis buffer
(LB; calcium/magnesium-free PBS containing 1% Nonidet P-40, 1
mM phenylmethylsulphonyl fluoride (PMSF), 1% aprotinin (Sigma),
1 mM EDTA and 10 mM sodium azide) and the post-nuclear fraction
separated on 10% polyacrylamide gels. Transfer and western blotting
was carried out as in Fig. 1 using appropriate affinity-purified
isotype specific goat anti-mouse alkaline phosphatase conjugates
(Zymed Inc., San Francisco). Microsomes were prepared from VG
cells by resuspending a washed cell pellet in 0.25 M sucrose containing
3 mM imidazole, PMSF and aprotinin, 1 mM MgCl2, 10
mM CaCl2 and passing four times through a ball-bearing
cell cracker (after W. Balch, Scripps institute, La Jolla, California)
followed by pelleting to 1 h at 100,000g in a Beckman TLA-45 rotor.
Microsomes were then subjected to carbonate washing 21
or proteinase K digestion. Proteinase K digestion was for 60 min
at 25 °C with 250 µg proteinase K ml-1 in
25 mM Tris-HCl buffer (pH 7.5) containing 10 mM CaCl2,
1 mM 1 mM dibucaine, 100 mM NaCl with or without 1% Triton X-100.
Digestion was stopped by the addition of 10 mM PMSF, precipitated
with an equal volume of trichloroacetic acid and dissolved in
reducing sample buffer before electrophoresis and transfer to
nitrocellulose. The 9D6 anti-idiotype secreting hybridoma cells
were lysed in LB, immuno-precipitated with goat anti-mouse IgG
followed by fixed Staph. A (Zysorbin; Zymed Inc.), washed
four times in PBS containing an additional 0.3 M sodium chloride
and 1% (v/v) Nonidet P-40, then washed once in water and eluted
into reducing sample buffer before electrophoresis and transfer
to nitrocellulose.
Radio-labelled antigen could be immunoprecipitated from [35S]methionine labelled lysates of 5D3 or 9D6 hybridoma cells (Fig. 2b) or from lysates of isolated microsomes (data not shown) simply by the addition of the appropriate second antibody and fixed Staphylococcus aureus cells. Pulse-chase analysis (data not shown) demonstrated that this complex existed intracellularly before cell lysis. The antigen immunoprecipitated with 5D3 was recognized in immunoblots by 9D6 and the antigen immunoprecipitated with 9D6 was recognized by 5D3 (Fig. 2b).
A third anti-idiotypic response was produced when 1D3 ascites was generated in unirradiated syngeneic mice. The ascites contained an IgM that was reactive with the same 72K band by western blotting (Fig. 2d, lane 5), but which was absent from 1D3 culture supernatant (Fig. 2d, lane 4). Presumably this is because the intact immune system of the tumour-bearing mice responds to the presence of high levels of 1D3 immunoglobulin by producing a primary (IgM) anti-idiotype response, which includes the internal image anti-idiotype specificity capable of recognizing the putative receptor. Taken together, these results indicate that three different routes of anti-idiotype production generate antibodies that recognize the same 72K conserved membrane protein.
The anti-idiotype antibodies recognize a common idiotope in antisera against KDEL peptides (Fig. 3). For example, 5D3 not only recognizes a rabbit antibody raised against the original KETE peptide but also rabbit antibodies raised against other KDEL peptides (Fig. 3a). The complementary assay (Fig. 3b) showed that the rabbit anti-KDEL antisera recognize 9D6 and 5D3 immunoglobulins more strongly than either the monoclonal antibody, 1D3, or another control antibody. No reactivity was seen against rabbit antibodies raised against two irrelevant peptides (Fig. 3a) or preimmune rabbit serum (Fig. 3a and b). This cross-specificity is particularly striking as some of the individual anti-tail antibodies show a much narrower range of reactivity. For example, a rabbit anti-KDDD antiserum that recognizes PDI does not recognize BiP, although rabbit anti-KEED, which recognizes BiP, does not recognize PDI (D.V., J.T. and S.F. manuscript in preparation).
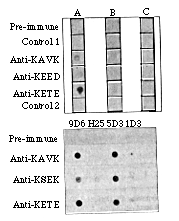
FIG. 3 The monoclonal anti-idiotype antibodies
recognize an idiotope in rabbit anti-KDEL antisera. Upper panel:
5D3 antibody (lane A) specificaliy recognizes rabbit antisera
to KDEL peptides, but not other rabbit antisera. This recognition
is not seen with other IgM monoclonal antibodies (lane B), nor
with second antibody alone (lane C). Lower panel: Rabbit anti-KDEL
antisera recognize both 5D3 and 9D6 immunoglobulin, but do not
react with other murine monoclonal antibodies (H25 and 1D3). This
effect is not seen with rabbit antisera to other antigens.
METHODS. Polyclonal rabbit antisera to the peptides in Table 1
were prepared35 and concentrated by ammonium sulphate
precipitation. For the upper panel, aliquots of concentrated rabbit
antisera (1 µl) were dried onto nitrocellulose filters which
were western blotted with culture supernatant of 5D3, culture
supernatant from a control IgM secreting hybridoma, or blocking
buffer and processed as described in Fig. 2. For the lower panel,
hybridoma culture supernatant from 5D3 or 9D6 or control hybridomas
was spotted onto nitrocellulose and probed with rabbit preimmune
serum or antisera to KDEL using the same blotting protocol. In
each case, the control antisera (Control 1 and Control 2) to irrelevant
peptides had titres on their cognate antigens comparable to those
of the anti-KDEL antisera on the immunizing peptides.
If the anti-idiotypes mimic the common structure of KDEL tails recognized by the receptor, then antibodies raised against 5D3 or 9D6 immunoglobulins should show the broad KDEL tail specificity of the receptor itself. This would also be proof that 5D3 and 9D6 are internal image anti idiotype antibodies. Evidence for this was obtained from a third round of immunization. Anti-5D3 immunoglobulin sera were generated in BALB/c mice by immunization with fixed 5D3 hybridoma cells. This antisera specifically recognized peptides terminated with KDEL and not those terminated with K...GL (Fig. 4 a). Furthermore, this recognition extended to KDEL peptides other than the original antigen. A comparable primary IgM response was found in the serum or ascites from nonirradiated mice bearing either anti-idiotype hybridoma as an ascitic tumour (Fig. 4b). These results demonstrate that anti-idiotypes elicited by 5D3 or 9D6 recognize the structural feature common to the KDEL tails. Hence, the protein identified by 5D3 and 9D6 should also recognize the KDEL signal on a variety of proteins, which makes it a good candidate for the role of KDEL receptor.
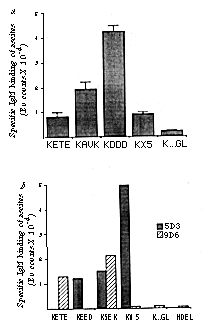
FIG. 4 Third-round immunizations generate anti-KDEL
speatic reactivity. a, Anti-5D3 antisera specifically recognize
KDEL-terminated peptides. b, Spontaneous IgM responses
in mice bearing anti-idiotype hybridoma tumours also show anti-KDEL
specificity.
METHODS. a, BALB/c mice were immunized four times, intraperitoneally,
with paraformaidehyde fixed 5D3 hybridoma cells (~5 x 106
cells per mouse) at intervals of three weeks and bled 10 days
after the final immunization. Sera were diluted 1:500 in DELFIA
blocking buffer and assayed in triplicate on KDEL and K...GL peptide
substrates by a solid-phase time resolved lanthanide fluorescence
assay36. Background values obtained with irrelevant
mouse antisera were subtracted to give specific binding. The assay
was performed three times using sera from three separate immunized
animals and a representative set of results is shown. b,
Ascites from unirradiated BALB/c mice carrying either 5D3 or 9D6
hybridoma tumours were clarified by ultracentrifugation and used
at dilutions of 1:100 in a solid-phase DELFIA assay in triplicate
on KDEL, K...GL and HDEL peptides. A highly purified affinity
isolated µ-chain-specific goat anti-mouse IgM second antibody
conjugated to a Europium chelate was used as detecting antibody36.
This antibody has negligible cross-reactivity with mouse IgG.
Although both antibodies were tested on all peptides, values below
2,000 Eu counts (photon counts of Europium fluorescence) are not
visible in this representation.
Anti-idiotype secreting hybridoma cells
The properties of the 5D3 and 9D6 cells themselves lend further
support to the assignment of the 72K antigen as the KDEL receptor.
We have already demonstrated that the antigen interacts with the
antibodies intracellularly, so it is not surprising that the hybridomas
grew poorly, cloned inefficiently and secreted only small amounts
of correctly assembled immunoglobulins. Nevertheless, the cells
survived and therefore must have compensated for the presence
of anti receptor antibody. There are two mechanisms by which antibody
interference with the retention of KDEL proteins could be overcome.
Either the KDEL ligands could be coordinately upregulated to maintain
functional levels in the lumen of the ER in the face of increased
leakage, or the receptor itself could be upregulated to titrate
the effect of the inhibiting antibody.
Analysis using a fluorescence-activated cell sorter (FACS) revealed that the anti-idiotype hybridomas exhibited the same steady-state levels of PDI, BiP and GRP 94 as hybridomas producing a variety of irrelevant immunoglobulins (shown for PDI in Fig. 5a-c). Pulse-chase analysis confirmed that the anti-idiotype hybridoma cells do not secrete large quantities of these normally retained KDEL proteins (data not shown). These results imply that the anti-idiotype cells did not overcome the retention defect by increased ligand expression. Instead, the hybridoma cells contained greatly elevated levels of the putative receptor. Both 5D3 and 9D6 cells contain the normal 72K membrane-associated protein and large quantities of a heterogenous soluble form (Fig. 2d). The soluble antigen-antibody complex was also found at high levels in media conditioned by the anti-idiotype hybridoma cells (Fig. 2b). The antigen in this material must have passed through the entire Golgi apparatus as a marked Mr shift was observed after neuraminidase treatment (data not shown). It is important to emphasize that soluble receptor is produced only by the two grossly abnormal anti-idiotype-secreting hybridoma cell lines. No other cell lines have been found that secrete material immunoreactive with the anti-idiotype antibodies.
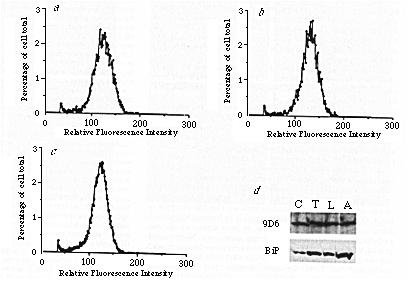
FIG. 5 a, b, c, PDI content
of rapidly growing SP2/0 myeloma cells (a), 5D3 hybridoma
cells (b) and VG hybridoma cells (c) is equal. d,
Expression of the 5D3/9D6 antigen is not rapidly upregulated as
part of the stress response.
METHODS. For a, b and c, cultures of SP2/0
myeloma cells and 5D3 and VG hybridoma cells in rapid growth were
washed twice in PBS, fixed in suspension in methanol at -20 °C
for 6 min, washed twice more in PBS and preincubated 1 h with
PBS containing 10% (v/v) goat serum and 10 mM sodium azide. The
fixed, permeabilized, cells were then incubated with 3 rabbit
anti PDI antiserum (Fig. 1) for 1 h, washed twice in PBS, reblocked
with the goat serum buffer and labelled for 1 h with a rhodamine-conjugated
affinity-purified goat anti-rabbit antibody37. The
cells were finally washed in PBS and analysed in an EPICS 4 (Coulter
Electronics) FACS. Profiles from 50,000 cells, taken from a representative
experiment are shown. For d, Vero cells were grown in normal
growth medium (lane C), medium containing 50 ng ml-1
tunicamycin (lane T), glucose-free medium (lane L), or medium
containing 0.5 µg ml-1 A23187 (lane A) for 4
h. Cells were then collected, lysed, and equal aliquots analysed
by SDS-PAGE and western blotted with 9D6 and a rabbit anti-BiP
antiserum (rabbit anti-KEED). The western blots were repeated
at several loadings of sample to ensure that neither second antibody
nor substrate were limiting. The 9D6 immunoblot shown contains
five times the protein-loading of the anti-BiP immunoblot.
The poor growth of the anti-idiotype-secreting hybridoma cells suggested that they might be under stress and prompted us to ask whether any of the observed up-regulation of antigen expression could be attributed to a stress response. In particular, as several of the soluble ER resident proteins are induced by glucose starvation, we tested this treatment for its ability to increase expression of the receptor. Figure 5d shows that the putative receptor was not upregulated under conditions that elicit strong induction of known glucose-regulated proteins. Furthermore, the putative receptor was not induced by treatment for up to 4 hours with the glycosylation inhibitor, tunicamycin, or the calcium ionophore, A23187 (Fig. 5d).
Soluble antigen binds to KDEL peptides and PDI
The soluble forms of the putative receptor, isolated from the
medium of overexpressing hybridoma cells, were first tested for
binding to KDEL-peptide affinity columns. The isolated receptor
bound specifically to the peptides containing the KDEL retention
signal, but not to K...GL (Fig. 6a). The observed efficiency
of binding was low (typically 3-5% of input counts), presumably
because this isolated material differs from the 72K transmembrane
receptor in normal cells; it is a soluble fragment, glycosylated
by passage through the Golgi apparatus and associated with the
anti idiotype antibody. Specific binding is obtained with material
isolated from either anti-idiotype hybridoma (Fig. 6a)
and is divalent cation-dependent and maximal at physiological
pH (data not shown). This indicates that divalent cations may
play a part in switching the receptor between high- and low-affinity
forms. The affinity of the unoccupied binding sites in the isolated
antigen for KDEL ligands was estimated by a complementary experiment.
This experiment measures the binding of radio-iodinated PDI to
isolated antigen attached to Sepharose beads (Fig. 6b).
Analysis of these data yields a dissociation constant Kd
of 23 µM. This binding can be competed by incubation with
KX5 peptide at a concentration of 100 µM, a value similar
to that calculated for KDEL tails in the ER lumen.
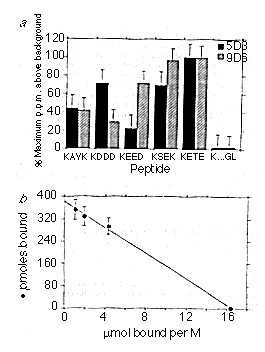
FIG. 6 Binding of isolated receptor to KDEL
peptides and PDI. a, Radiolabelled soluble receptor isolated from
either anti-idiotype hybridoma cell line binds to several KDEL
peptides, but not to the K . . . GL peptide. b, lodinated PDI
binds specifically to soluble antigen immobilized on Sepharose
beads.
METHODS. 5D3 or 9D6 cells were labelled for 14 h with [35S]methionine
and the soluble fragment of the receptor isolated from the supernatant
by chromatography on concanavalin A-Sepharose (Pharmacia) according
to manufacturers instructions, followed by dialysis against TN
containing 1 mM dithiothreitol. a, Peptides were crosslinked
to CnBr Sepharose (Pharmacia) according to manufacturer's instructions.
The peptide-Sepharose particles (50 µl of a l:l v/v slurry)
were preblocked by incubation at room temperature in 1 ml blocking
buffer (TN 2% Tween-20, 1 mM CaCl2, 1 mM MgCl2,
20% FCS) for 2 h with gentle agitation, then incubated with constant
aliquots of the labelled receptor overnight at room temperature.
The peptide-Sepharose was then washed three times in blocking
buffer without FCS and counted in a liquid scintillation spectrometer.
The results shown are the average of four experiments, with each
point in triplicate. Error bars indicate the standard error. Background
corresponding to beads alone has been subtracted. In a typical
experiment, the background value was 650 counts and the highest
sample value was 1,800 counts or 3-5% of input counts. All values
except K . . . GL and KEED (5D3) are significantly above background
(P < 0.05). Gel electrophoresis of a larger quantity of KSEK-Sepharose
after incubation with isolated 5D3 receptor showed that the only
radio labelled protein bound co migrated with the soluble form
of the receptor. b, PDI was isolated from bovine liver
by the method of Lambert and Freedman32 as modified
in Tooze et al.32, PDI was iodinated using lodobeads
(Pierce) according to manufacturer's instructions at a concentration
of 5 µg ml-1 and separated from free label by
chromatography on Sepharose G-50. The specific activity of the
labelled PDi was 1.05 x 107 c.p.m. µg-1
(0.18 atoms iodine per PDI tail) 9D6 soluble antigen was isolated
as above and coupled to cyanogen bromide-activated Sepharose 4B
according to manufacturer's instructions at a concentration of
~5 mg per ml of beads. For the binding assay, beads were pre blocked
by incubation with Blotto containing 1 mM DTT 1 mM CaCl2
and 1 mM MgCl2 for more than 1 h at room temperature
and then incubated 1 h at room temperature with 35,000 c.p.m.
ml-1 of labelled PDI in the same buffer, together with
unlabelled PDI at concentrations varying between 0 and 3 mM. Beads
were then pelleted and the percentage of free counts calculated.
Results of a representative experiment carried out in triplicate
are shown as means and standard deviation. The line indicates
a Kd of 23 µM, and has a correlation coefficient
of 0.9986. In four independent experiments Kds of 20-50
µM were found. Control experiments with beads alone showed
a Kd greater than 10 mM.
Localization of anti-idiotype reactivity
Immunofluorescence with 5D3 on BHK-21 cells maintained at
37 °C (Fig. 7a) yielded a weak punctate staining pattern
throughout the cytoplasm, which tended to concentrate in a crescent
next to the nucleus. We reasoned that the low level of signal
might reflect the fact that the receptor is almost continuously
occupied during the normal functioning of the secretory pathway.
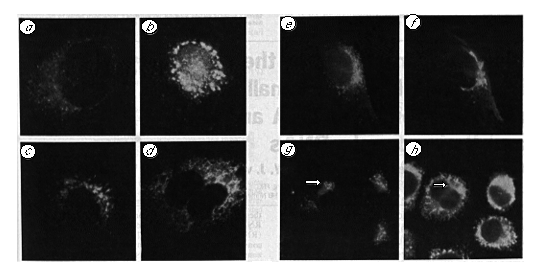
FIG 7 Intracellular location of the receptor.
Methanol-fixed NRK cells were labelled with 5D3 culture supernatant
after incubation at 37 °C (a), 2 h at 15°C (b),
2 h at 37 °C in the presence of 10 µg ml-1
brefeldin A (c), after 30 min recovery at 37 °C from
a 2 h brefeldin A block (d). e, Labelling of NRK
cells with mouse polyclonal anti-receptor antiserum. f,
The same cell labelled with a rabbit polyclonal antiserum against
the Golgi apparatus. g Labelling of methanol-fixed untreated
NRK cells with an affinity-purified rabbit polyclonal antiserum
to the soluble receptor fragment. The same field shown in (h)
double labelled with the mouse monoclonal antibody, 1D3, to reveal
the distribution of PDI. The arrow indicates a region of a flattened
cell in which receptor is concentrated in an area from which PDI
is excluded.
METHODS For immunofluorescence NRK cells were grown on multiwell
slides, treated as described above, washed briefly in PBS of the
appropriate temperature and fixed in methanol at -20 °C for
6 min. The cells were washed twice in PBS and incubated in blocking
buffer (PBS containing 10% (v/v) normal goat serum) at room temperature
for 30 min in a humidified chamber. First antibody incubations
were for 1 h at room temperature. The slides were washed five
times with PBS, re-blocked with blocking buffer then exposed to
fluorochrome-conjugated second antibody (Zymed Inc.) diluted according
to the manufacturer's instructions in blocking buffer for 1 h.
After extensive washing in PBS, the slides were mounted with Moviol
and examined in a Leitz Axiophot microscope. Photography was with
Tri-X rated at 1,600 ASA. Final magnification, X6,300 (all panels).
Treatments that interfere with the secretory pathway might be expected to interfere with recycling to the ER and result in decreased receptor-occupancy and therefore increased reactivity with the antiidiotype antibodies. Incubation at 15°C blocks transport between ER and Golgi and causes elaboration of a novel tubular structure in the peri-Golgi region14. Incubation of BHK-21 cells at 15 °C for 2 hours caused an increase in the intensity of the immunofluorescence obtained after staining with 5D3, together with a tendency for the punctate stain to coalesce into larger strongly labelled regions in the cytoplasm (Fig. 7b). Under the same conditions, anti-KDEL antibodies gave their normal reticular pattern of immunofluorescence (data not shown). Hence, incubation at 15 °C, a treatment that interferes with ER to Golgi transport14, increases the accessibility of the 72K putative receptor to specific anti-idiotype antibodies and alters its localization. This observation associates the receptor with the salvage compartment14.
Brefeldin A has a profound reversible effect on the exocytic pathway, which is manifest as a condensation of cis and medial Golgi into the ER24-25. When NRK cells were treated with brefeldin A for 2 hours the normal Golgi structure was completely dispersed, although the reticular pattern associated with the ER labelling of rabbit anti-KDEL antisera remained essentially unchanged. Under these conditions staining with 5D3 revealed the usual vesicular pattern, but with increased intensity (Fig. 7c). After removal of the brefeldin A, 5D3 showed a reticular ER-like pattern of labelling (Fig. 7d) before returning to the normal staining pattern seen in Fig. 7a. This behaviour also associates the antigen with the salvage compartment25.
Polyclonal antisera, raised in BALB/c mice by immunization with soluble receptor from either 5D3 or 9D6 cells, labelled the 72K band in VG cell lysates (Fig. 2d). These polyclonal antisera showed an immunofluorescent labelling pattern that was indistinguishable from the pattern obtained with 5D3. The widely distributed punctate labelling seen with these reagents showed the same tendency to concentrate to one side of the nucleus (Fig. 7e) and the same appearance in a juxtanuclear tubular compartment after a 15 °C block (data not shown) as observed with the anti idiotype antibody. Staining of the field shown in Fig. 7e with a polyclonal antiserum against the Golgi apparatus gives an overlapping but quite distinct cisternal pattern (Fig. 7f). Double-label immunofluorescence with the polyclonal antiserum to soluble receptor fragment (Fig. 7g) and I D3 (Fig. 7 h ) show that the receptor and its ligand have overlapping but distinct locations.
Discussion
The anti-idiotypic approach to the identification of a receptor
for the KDEL retention signal rests on the assumption that a single
receptor is responsible for the recognition of the several KDEL
proteins. Consistent with this assumption, three independent approaches
led to an anti-idiotypic response against KDEL peptide antibodies
and these independently-produced antiidiotypic antibodies all
recognized the same 72K transmembrane protein. The third round
of immunization provided the strongest support for our initial
assumption that one receptor serves several KDEL terminated ligands.
Antisera produced in response to immunization with 5D3 cells show
a broad specificity for the KDEL signal but do not recognize the
nonfunctional K . .. GL tail. A similar effect can be seen in
the third-round response generated by production of ascites in
non-irradiated mice from either of the anti-idiotypic hybridomas.
These responses again show reactivity with several KDEL tail sequences
but not with K . . . GL or HDEL peptides. This reactivity justifies
the identification of the 72K antigen as a KDEL signal-binding
protein and our localization and functional studies are consistent
with a role in recognition of the KDEL retention signal in
vivo. Henceforth we will refer to the antigen as the receptor.
Both 5D3 and 9D6 hybridomas secrete in large amounts a soluble form of the receptor that is highly glycosylated, oligomeric and complexed with antibody. Labelling experiments show that the production of soluble receptor exceeds antibody production in both hybridomas. Polyclonal antibodies against the secreted soluble receptor from 9D6 and 5D3 cells recognize the normal 72K receptor in VG cells. Therefore the secreted forms share antigenic determinants with the normal receptor, beyond those recognized by 5D3 and 9D6 antibodies. Whether the soluble form results from changes in transcriptional or post-transcriptional events remains to be seen, but we interpret overproduction of soluble receptor as a mechanism to titrate the anti-receptor antibodies in the lumen of the ER. Intact membrane-bound receptor present in the hybridoma cells and protected in this way from the antibody can then function sufficiently to permit survival of the cells.
The epitope recognized by 5D3 and 9D6 seems to be widely conserved throughout evolution. All of the vertebrate cell lines and tissues tested that show reactivity with our anti-tail antibodies (S.F., J.T. and D.V., manuscript in preparation) also react with the anti-idiotype antibodies. None of the other cell lines tested showed the overexpression and secretion of the antigen observed in the 5D3 and 9D6 hybridomas. In normal cells the receptor seems to be a minor membrane protein. Our anti-KDEL tail antibodies show negligible reactivity with a peptide bearing the S. cerevisiae signal (HDEL) or with cell lysates from this yeast. Correspondingly, our anti-idiotype antibodies show no significant reactivity in this organism.
Preliminary studies (results not shown) provide some intriguing insights about the receptor. Western blotting with a battery of our anti-KDEL tail antibodies indicate that the receptor is not itself a KDEL protein. The receptor is an intrinsic membrane protein that spans the membrane at least once (we cannot exclude several membrane-spanning domains). Nonreduced gels of VG lysates indicate the protein may be oligomeric and the antigen precipitated from 9D6 or 5D3 cells is disulphidecrosslinked to form apparent trimers and higher-order oligomers. Finally, the secreted receptor is posttranslationally modified. That the secreted antigens from 5D3 and 9D6 cells, although cross-reactive, migrate at different Mrs reflects their sialylation, as this difference in mobility is abolished by neuraminidase-digestion. The receptor in normal cells does not show this high degree of glycosylation, presumably because it is not exposed to the later stages of the secretory pathway.
The localization of the receptor is central to an understanding of its function in the cell. The patterns of immunofluorescence labelling of NRK cells at 37°C with 5D3, which should be specific for receptor that does not have KDEL ligand bound to it, indicate that the receptor is in a compartment distinct from the ER and the Golgi apparatus but related to them both. The labelling of cells incubated at 15° C establishes that the receptor is in the 15° C compartment, which we equate with the salvage compartment. The patterns of labelling during brefeldin A treatment and recovery from it are also consistent with the proposal that unoccupied receptor is in a compartment between the ER and the Golgi stack, that is, the salvage compartment. Here, the unoccupied receptor is positioned appropriately to capture KDEL proteins that escape from the ER and to recycle them. Results obtained with polyclonal and monoclonal antibodies against isolated receptor show a very similar labelling pattern, suggesting that only low levels of receptor are ever found in other cellular compartments. We intend to investigate the localization of the receptor by EM immunocytochemistry.
The binding of a recycling receptor to KDEL tails must be modulatable. The receptor must have a high affinity for KDEL tails when it captures them and a lower affinity under the conditions of the ER where they are released. The secretion of a soluble form by the hybridomas allowed us to isolate receptor for binding studies. Binding of this material to peptide columns demonstrated that it specifically recognized the KDEL terminated peptides rather than a K... GL peptide. This specific binding was strongest near physiological pH and was divalent cation-dependent. Pure labelled PDI bound to isolated receptor with a Kd of ~23 µM. Although post translational modifications of the secreted receptor may cause a change in the characteristics of binding, this Kd is within the physiological range. The concentration of KDEL tails calculated for the ER lumen is well above this constant, indicating that our binding protein is a plausible candidate for the physiological receptor. In addition, this value suggests that the difference between the high-affinity (salvage compartment) and low-affinity (ER) forms need not be large. The transferrin receptor, for example, has only a 20-fold greater affinity for diferritransferrin than for apotransferrin26. Such a change in the affinity of our putative receptor would allow it to function in the recycling and release of KDEL proteins into the ER lumen, where their concentration is high. The size of this affinity constant also provides an explanation for the difficulty experienced by us, and others, in isolating a KDEL receptor by conventional affinity methods. Finally, the conclusion that receptor binding is divalent cation dependent is supported by recent work27 that suggests the involvement of Ca2+ in the retention of KDEL-terminated proteins in the ER in vivo. Furthermore, we have observed that the availability of cellular receptor for binding to anti-idiotypic antibody in immunofluorescence experiments decreased after incubation of cells in medium containing the calcium ionophore A23187 and Ca2+ (S.F., J.T. and D.V., manuscript in preparation).
This work has identified a KDEL binding protein that we think
is a good candidate for the physiological receptor. In summary,
our evidence that the 72K protein is indeed the receptor comprises
the following: (1) separate routes beginning with different KDEL
sequences identify the same oligomeric transmembrane antigen;
(2) a broadened specificity is observed in the third-round antibodies;
(3) the antigen is concentrated in a compartment intermediate
between ER and Golgi; (4) the KDEL binding site lies in the lumen
of the secretory pathway; (5) the anti-idiotype hybridomas dramatically
upregulate this protein; (6) isolated antigen binds specifically
to peptides containing the retention signal and (7) isolated antigen
binds physiological ligand with a Kd compatible with
a recycling model for ER retention. Although it remains formally
possible that the localization and binding properties of this
protein are only coincidentally those expected of a KDEL receptor,
the properties of this protein are consistent with the model of
Munro and Pelham10, and indicate additional features
of ER retention. The binding site of one receptor species can
accommodate all known KDEL signals, hence several receptors are
not necessary. The receptor is a minor transmembrane protein,
which must therefore operate by a recycling mechanism. The receptor
is not normally detectable within the bulk of the ER or the Golgi
apparatus. The receptor appears restricted to a relatively small
region that may represent the exit sites from the ER and the return
sites for captured ligands. Both 15°C incubation and brefeldin
A treatment cause changes in receptor localization consistent
with it being predominantly present in the intermediate or salvage
compartment. The observed Kd of receptor for ligand
would allow the receptor to capture ligand in the salvage compartment
and release it into the ER without requiring large changes in
affinity constant. The moderate changes necessary could be mediated
by an environmental factor such as pH or divalent cation concentration.
Whether the receptor recycles continuously or only when occupied
by ligand is unclear; but we note that not only is the receptor
a transmembrane protein but it and several of its ligands are
oligomeric3. This opens the possibility that the receptor
signals its occupancy to the cytoplasm by clustering28.
Received 22 December 1989: accepted 27 April 1990.
1. Blobel, G. & Dobberstein, B. J Cell Biol. 67,
852-B62 (1975).
2. Palade, G. Science 189, 347-358 (1975).
3. Pelham, H R. B. A. Rev. Cell Biol. 5,1-24 (1989)
4. Warren, G. Nature 327, 17-18 (1987).
5. Haas, I. G. & Wabl, M Nature 306, 387-389 (1983)
6. Freedman, R B. & Hillson, D. A Enzymology of Post-translational
Modification of Proteins157-212 (Academic, London, 1980).
7. Freedman, R. B Trends Biochem. Sci. 9, 438-441(1984),
8. Weibel, E. R., Staubh, W., Gnagi, H. R & Hess, F A J.
Cell Biol. 42, 68-91(1969).
9. Munro. S. & Pelham, H R B. Cell 46, 291-300 (1986)
10. Munro, S & Pelham, H. R. B. Cell 4S, 899-907 (1987)
11. Pelham, H R. B., Hardwich, K. G & Lewis, M J. Embo
J. 7,1757-1762 (1988)
12. Ceriotti, A & Colman, A. EMBO J. 7, 633-638 (1983).
13 Pelham H. R B. EMBO J. 7, 913-9l8 (1988)
14 Saraste, J. & Kuismanen. E. Cell 38, 535-549 (1984)
15. Tooze. J., Tooze, S. & Warren. G Eur J. Cell Biol.
33, 2B1 293 (1984)
16. Balch W E, Elliott, M. E. & KeMer, D S. J. Biol. Chem.
261,14681-14689 (1986).
17. Gaulton, G. N & Greene, M. L. A. Rev. Immun. 4,
253-280 (1986).
18. Vaux, D J. T, Helenius, A & Mellman, I. Nature
332, 36-42 (1988).
19 Farid. N R. & Lo, T C. Y. Endocr. Rev. 6,1-23 (1985).
20 Pain, D., Kanwar, Y S. & Blöbel, G. Nature
331, 232-237 (1988).
21 Tooze, J, Kern. H F, Fuller. S D & Howell. K E J. Cell
Biol. 109, 35-50 (1989).
22. Smith, M. J. & Koch, G. L. E. EMBO J. S, 3581-3586
(1989).
23 Bordier, C. J. Biol. Chem. 256,1604-1607 (1981).
24. Misumi, Y. et al. J. Biol. Chem. 261,11398-11403 (1986).
25. Lippincott-Schwertz, J. et al. Cell 60, 821-836 (1990).
26. Klausner, R. D. et al. Proc. Natn. Acad. Sci. USA.
S0, 2263-2266 (1983). 27 Booth, C. &
Koch, G. L. E. Cell 59, 729-737 (1989).
28 Fulier, S. D Cell 45, 923-934 (1987).
29 Frank, R. & Gausepohl, H. in Modern Methods in Protein
Chemistry Vol. 3 (ed. Tschesche, H ) 42-60 (De Gruyter, Berlin
& New York).
30. Laemmli, U. K. Nature 227, 680-685 (1970).
31. Johnson, D. A. et al. Gene Analyt. Techn. 1, 3-8 (1984).
32. Lambert, N. & Freedman, R. B. Biochem. 1213, 225-234
(1983).
33. Fisher. P et al. J. Cell Biol. 92, 674-686 (1982).
34 Fazekas de St Groth, S. & Scheidegger, D. J. Immun.
Methods 35,1-21 (198O).
35 Kreis T. EMBO J. 15, 931-941 (1986).
36. Hemmila, I. et al. Analyt. Biochem. 137, 335-343 (1984).
37. Vaux, D J T & Gordon, S. J. Cell Sci. 77,109-127
(1985).
ACKNOWLEDGEMENTS. We acknowledge the technical assistance and forebearance of P Buck and M. Kail. We thank our colleagues R. Frank and H. Gausepohl for the synthesis of the peptides and for useful advice on peptide chemistry. We thank B. Burke for the gift of a polyclonal antiserum against a protein of the Golgi apparatus. We also thank P. Walter and H. Pelham for their comments and encouragement.
![]()
![]()
© Copyright 2000 Department
of Biology, Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu