The web page is segregated into three different sections for the KGD1 protein and the hypothetical YIL127C protein. The first section discusses protein structure. The second section discusses protein interactions. The third section outlines experiments that may yield quantitative and qualitative information in regards to protein structure and interactions.
KGD1
From my previous web pages (project 2, project 3) we know the following information about KGD1: it is the E1 subunit of the alpha-ketogluterate dehydrogenase complex, KGD1 encodes the protein 2-oxoglutarate dehydrogenase which is located in the mitochondria matrix, KGD1 protein is approximately 1014 amino acids long, corresponding to 114 kda (SwissProt) for the unprocessed precursor, and 2-oxogluterate dehydrogeanse is part of three different metabolic pathways: the TCA cycle, tyrptophan metabolism, and lysine degradation.
STRUCTURE
Figure 1: PDB file for yeast Lipoamide Dehydrogenase (E3) chains A and B which is part of the 2-oxogluterate dehydrogenase complex. The PDB database does not currently have a structure for KGD1 (2-oxogluterate dehydrogenase) or the alpha-ketogluterate dehydrogenase protein complex.
From the What is There database (WIT) the following link (click here) lists KGD1 as having a Mg2+ ion cofactor and a Thiamine diphosphate coenzyme (figure 2).
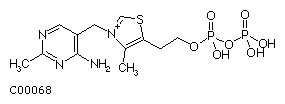
Figure 2: Structural drawing for thiamine diphosphate (TPP).
ScanProsite lists potential protein domains for post translational modifications for KGD1. (All of the following hits are for amino acid sequences with a high probability of occurrence.)
| Post-translational modification |
Number of Sites: |
N-glycosylaytion site |
8 |
cAMP and cGMP-dependent protein kinase phosphorylation
site |
1 |
Protein kinase C phosphorylation site |
18 |
Casein kinase II phosphorylation site |
23 |
N-myristoylation site |
10 |
Table 1: Predicted post translational modification sites for the 2-oxogluterate dehydrogenase E1 component of the alpha-ketogluterate dehydrogenase complex. The majority (76.7%) of post-translational modifications are for phosphorylation. There are also three different phosphorylation mechanisms, which indicate that the protein may be deactivated or hyperactived in three different signaling cascades. One potential glycosylation site and one potential myristoylation site may be responsible for protein localization.
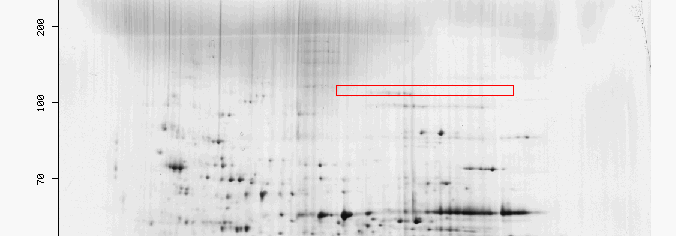
Figure 3: The red box highlights the region were 2-oxogluterate dehydrogenase would be found in a two 2-D gel for yeast. It is unusual that there are very few spots in the box highlighted for KGD1 since KGD1 is an enzyme prevalent in three different metabolic pathways. Two possibilities may explain this phenomenon. First, KGD1 protein may still be bound in the alpha-ketogluterate dehydrogenase complex which has a different pI, and molecular weight. Second, if the yeast utilized for this 2-D gel were grown in conditions where KGD1 was not highly expressed and translated, then the protein would be present in small quantities.
PROTEIN INTERACTIONS:
Figure 4: DIP protein interactions for KGD1. The node in the center (colored pink) is KGD1. Table 2 lists some of KGD1's nearest neighbors. There are two trends for the dip graph. First, KGD1 interacts with many different proteins. Second, most of these interactions appear to be weak. (The legend in the bottom right corner indicates the confidence level between two node interactions. Low confidence in the interaction is denoted by a thin edge, high confidence is denoted by a thick edge.)
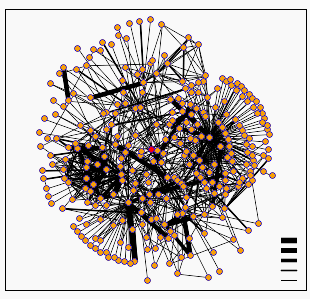
Table 2: Nearest nodes to KGD1. There does not appear to be a trend for KGD1 and its nearest neighbors in terms of function or localization. The nearest neighbors also do not contain KGD2 or LPD2 proteins which are known to interact with KGD1 protein to form the alpha-ketogluterate dehydrogenase complex.
Gene |
Corresponding protein/ function or localization
or keywords associated with the protein |
KGDP1 |
Phosphfructokinase 1/ ATP, glycolysis; phosphotransferase,
|
IMB3 |
Karyopherin beta-3 subunit, protein secretion enhancer
I/ transmembrane protein involved in ribosome transport |
IF4A |
Eukaryotic initiation factor 4A/ ATP; DEAD box;
P-loop; protein biosynthesis; purine nucleotide binding; RNA binding
|
YM24 |
Hypothetical protein |
SERA |
D-3-phosphoglycerate dehydrogenase I/ oxidoreductase;
serine biosynthesis, utilizes NAD+ |
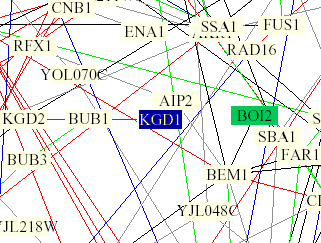
Figure 5: Yeast interaction map from Benno Figure 1. KGD1 is in the center of the image and highlighted blue. KGD1 interacts with BUB1 by a single red edge indicating the proteins have the same subcellular location and cellular role. KGD1 is also highlighted in a light yellow box indicating that the protein is involved in lipid metabolism. BUB1 has the following molecular function, "INVOLVED IN CELL CYCLE CHECKPOINT ENFORCEMENT. CATALYZES THE PHOSPHORYLATION OF BUB3 AND ITS AUTOPHOSPHORYLATION" and has a nuclear subcellular location (SwissProt). These results are unusual because I would not expect a protein with a mitochondrial and metabolic function to be linked with a nuclear phosphorylating protein. It should be noted that the model was incorrect 28% of the time (Campbell et al., 2002). However, in lu of the data from Scanprosite, KGD1 might be phosphorylated at some point during the yeasts life.
Databases yielding no significant hits.
The protein had to be truncated by at least 214 amino acids in order for the server to preform ccomputations on protein folding for KGD1 protein. Therefore, I did not include the results because I didn't know how the truncation would affect the folding patterns for KGD1, the catalytic activity of KGD1, and KGD1's ability to trimerize with KGD2 and LPD1 proteins.
Y2H Zero hits for KGD1
Yeast Path Calling Zero hits for KGD1
Triples one insertion for an mTn transposon, but the insertion did not result in a new phenotype or proposal of a non-annotated open reading frame (NORF).
Conclusions:
The data suggest two important trends. First, the data from Benno figure 1
and scanprosite suggest that KGD1 may be phosphorylated at some point during
the yeast's cell cycle. This may either hyper-activate or deactivate the protein
in a metabolic pathway. It should be noted the domains for post tranlsational
modification were listed as having a high probability of occurrence and the
Benno figure was incorrect 28% of the time. Second, the Benno figure illustrates
that circuit diagrams for protein interactions are still in their infancy.
It is difficult to represent a dynamic proteome in a static picture.
As an aside, I was troubled by the fact that there was not an abundance of
KGD1 protein in the 2-D gel. I investigated the plausibility that the alpha-ketogluterate
dehydrogenase complex contained a transmembrane domain which would render
it insoluble during the 2-D gel preparation. A kyte-doolittle hydrophobicity
plot indicates that LPD1 has two transmembrane domains (see figure 3), suggesting
that the alpha-ketogluterate dehydrogenase complex may be anchored to the
cristae of the mitochondria. This hypothesis assumes that the binding in the
quaternary structure of the alpha-ketogluterate dehydrogenase complex is strong
and not susceptible to disassociation in aqueous solution. With this information,
it may be possible to isolate the alpha-ketogluterate complex for a 2-D gel
analysis by using a detergent with similar properties to Triton X-114
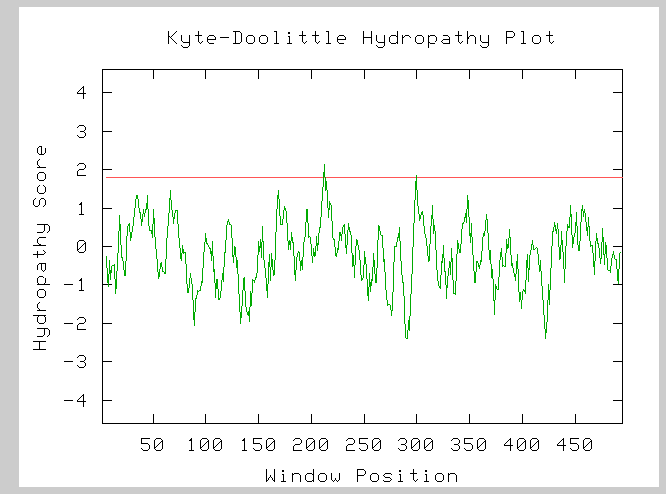
Figure (7): Hydorphobicity plot for the LPD1 subunit of the alpha-ketogluterate dehydrogenase complex. The two peaks which cross the red line at 1.8 on the hydropathy axis indicate two possible transmembrane domains.
Experiments.
The protein structural and interaction information from the databases suggest three different sets of future experiments. For the first set of experiments, I would test two different hypotheses. First, is KGD1 phosphorylated? To answer this question, I would utilize Chait’s et al. (Campbell, 2002) stable isotopes method. I would take sample yeast from cell populations grown on N15 and N14 media at various time points during the yeast’s life cycle. Using Maldi MS/MS I would analyze different peak ratios for large differences. I would predict that at some point during the yeast’s life KGD1 is phosphorylated to either deactivate or hyperactivate KGD1 proteins for a particular metabolic pathway. Phosphorylation may correspond to increased energy needs during sporulation or cell division. As an extension to this experiment I would utilize the ICAT method developed by Abersold et al. (Campbell, 2002) to find what growth conditions result in a preponderance of phosphorylated KGD1 protein. For the second set of experiments, I would test the strength of the quaternary interactions for KGD1, LDP1 and KGD2. This experiment would probably be done with classical biochemical approaches such as slowly increasing the strength of detergent washes and measuring the disassociation constant for the alpha- ketogluterate dehydrogenase complex with fluorescent probes. I could also measure the disassociation constant for different aqueous solutions (high salt, low salt, increased and decreased pH). I would predict that the quaternary interactions are moderate to strong. Site directed mutagenesis experiments would indicate which amino acid residues are responsible for quaternary structure in the alpha-ketogluterate dehydrogenase complex. Another approach to testing these interactions is to take crystals for LPD1 protein and incubate them with KGD1 and KGD2 proteins to determine whether or not the LPD1 proteins can be used as seed crystals for the formation of the alpha-ketogluterate dehydrogenase complex. If this experiment works, then X-ray crystallography could be used to analyze the alpha-ketogluterate dehydrogenase complex. For the third set of experiments, I would study the metabolic properties of KGD1. Employing Diovich’s et al. (Campbell, 2002) techniques for studying zeptomoles of a particular protein, I would preform two different experiments. The first experiment would test the catalytic ability of single KGD1 proteins for three different metabolic substrates, since it is involved in three different metabolic pathways. The second experiment would be an extension of the first and would determine how phosphorylation affects the maximum velocity (reaction rate) of single enzymes. The third experiment would use metabolites as antigens on an antigen microarray to confirm KGD1’s involvement in three different metabolic yeast pathways.
YIL127C
YIL127C is a hypothetical 23.8 kda protein with 206 amino acids. Sequence and structural databases predicts that YIL127C interacts with an ATPase. DNA microarray analysis, however, suggests that YIL127C is involved in RNA processing, RNA binding and has a sub cellular location in the nucleus. Keep in mind that a Kyte-Doolittle hydrophobicity plot indicates YIL127C protein had no transmembrane domains. Predator predicts that YIL127C protein was primarily random coils (53.47%)
Structure:
ExPasy: For YIL127C the hypothetical pI is 9.83 and the MW is 23845.14 da. YIL127C would be found in the region highlighted in figure 5.

Figure 8: 2-D gel for yeast. This highlighted region has a few spots which may or may not correspond to YIL127C.
YIL127C is classified as an unstable protein with an estimated half life of >20 hours. These results seem contradictory since an unstable protein is more likely to have a short half life, than a long half life. Nevertheless, if the half life prediction is correct, then the protein for YIL127C should be present for a significant amount of time during the the life of a yeast.
This database predicts YIL127C will have a subcellular location in: the cytoplasm (64%), nucleus (25%), and the mitochondria (10%). This data is consistent with are hydrophobicity plot which indicated that YIL127C does not have transmembrane domains and is a soluble protein.
Databases yielding no significant results:
ScanProsite: YIL127C had zero hits when screen was applied for domains with a high probability of occurrence
3D-Pssm: E-values were high, indicating a lack of similarity between fold models and YIL127C
Triples: No insertion or NORF is reported for YIL127C.
Yeast Path Calling: No interactions were listed for YIL127C in this older database
Dip: The graph for protein interactions was unavailable
Benno Figure 1: YIL127C was not present in the figure
Y2H: Zero hits for YIL127C
WIT: Zero hits for YIL127C
Conclusions:
Many databases did not have any proetomic information for YIL127C and it is difficult to make any predictions concerning the structure and interactions of YIL127C protein other than those that have already been made in my previous web pages (project 2, project 3). The most significant finding is from the Yale Geirstein database which predicted that YIL127C protein would localize in the cytoplasm. Hence, the best place to isolate and quantify YIL127C may be from the cytosal.
Experiments:
The key to uncovering the mystery of YIL127C is to quantify and isolate the protein. In order to accomplish this I would use size exclusion chromatography to isolate fractions of yeast cytosal proteins with MW small than 30 kda. Using a pI column I would separate each protein in preparation for a 2-D gel. I would then look in the predicted region for small spots, isolate each spot and sequence the amino acids. Because YIL127C is predicted to be an unstable protein and is probably not abundant, I would predict it would be a difficult and tedious task to isolate YIL127C protein. However, by analyzing DNA micrroarry experiments in which YIL127C induction is high, it might be possible to isolate yeast that have a higher concentration of YIL127C. This would make identification and quantification slightly easier in a 2-D gel.
Furthermore, once YIL127C protein is isolated, I would use an antigen microarray experiment to test whether or not the protein has any DNA or RNA binding capabilities. The antigen microarray would use genomic representations as antigens to determine whether YIL127C protein bound to DNA and where it bound. Assuming YIL127C bound to DNA, an antigen microarray with oligomers overlapping every 10 bp could be used to isolate the specific sequence YIL127C protein bound to. This technique could also be applied to RNA, although it would be difficult since RNA is unstable and susceptible to degradation. Proteomic and mirrcroarray data suggest that YIL127C protein may bind to mRNA in the cytosol. Thus, I would predicte that YIL127C binds to RNA and may have a post-transciprtional molecular function. The antigen micrroarray experiments should be carefully controlled to prevent generating false positives.
To test for protein interactions, I would utilize the yeast two hybridization method to test whether or not YIL127C protein binds to proteins associated with mRNA processing the cytosol. I would predict that if YIL127C binds to mRNA, then it might bind to proteins associated with mRNA processing. Therefore, another possible function of YIL127C may be to act as an intermediary and help specific proteins bind to mRNA.
References:
1. Campbell, M., Heyer L. Dicovery Genomics Proteomics & Bioinformatics. Cold Spring Harbor Labratory and Benjamin Cummings. San Francisco. 2003.
2. Schwikowski, Benno, Uetz P., Fields S., A network of protein-protein interactions in yeast. Nature Biotechnology. 18: 1257-1261.
Davidson College Biology Home Page
© Copyright 2002 Department of Biology, Davidson College, Davidson, NC 28035
Send comments, questions, and suggestions to: alcubre@davidson.edu