*This web page was produced as an assignment for an undergraduate course at Davidson College*
My Favorite Yeast Genes' Expression Patterns
(back)
Vas1 and YGR093w, my favorite chromosomal neighbors
Introduction
Public microarray databases are troves of scientific data. Much, if not most, of that information remains unexplored. As part of the public domain, these databases may contain readily available answers to a number of research questions. On this webpage, I will utilize yeast microarray database websites to examine the expression patterns of my favorite yeast genes: Vas1 and YGR093w. The former is a known gene. I will compare the expression profiles I find to my expectations about its transcription based on my knowledge of its biological process and molecular function. YGR093w is a predicted ORF with no known molecular function or biological process. Analysis of YGR093w's expression profile will allow me to test and refine my hypothesis about its function.
Microarray Data
DNA microarray analysis provides a powerful new method of biological research. In order to fully utilize the data produced by microarrays, math, biology, and computer science must all be combined. This developing, hybrid discipline is known as Bioinformatics. Microarrays allow researchers to quantify the amount of expression of every known or predicted ORF in any sequenced genome. A microarray is an organized collection of "spots". Each spot represents a single probe. This probe may be an oligonucleotide or PCR product. The probe will bind a specific piece of DNA or mRNA if it is present. This provides a quantitative measure of gene expression. While the number of spots is limited, it is currently possible to analyze the expression of every known human gene at once on a single microarray slide. More than simple quantification of expression, however, the most powerful application of this technology has been comparison of expression levels under varying experimental conditions.
In a simple microarray experiment, mRNA is collected from cells under two different conditions. The control population would be under "normal" conditions and the experimental population of cells might be exposed to anything from heat shock to glucose limitation. For a more detailed description of microarray methodology, try this introductory Flash animation (DNA Microarray Methodology, 2004 <http://occawlonline.pearsoned.com/bookbind/pubbooks/bc_mcampbell_genomics_1/medialib/method/chip/chip.html>). By convention, mRNA from the control population is dyed green while experimental mRNA is dyed red. When the two colors are scanned and displayed on top of each other, data comes in the form of colored spots (Figure 1).
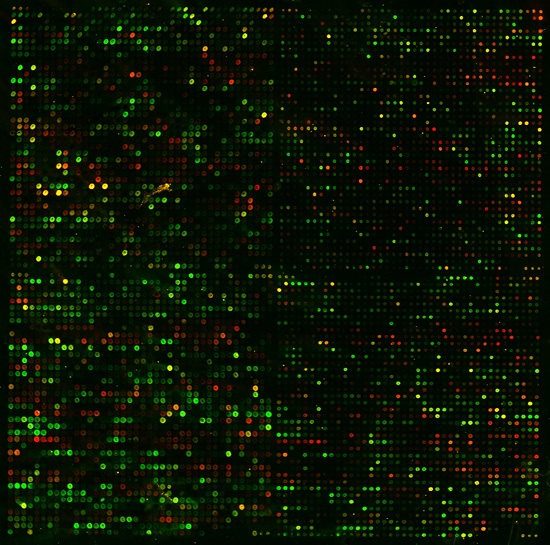
Figure 1. Permission pending from Dr. Patrick O. Brown. The results of a sample yeast microarray experiment is captured above. Each spot represents a single yeast gene. The color of each spot corresponds to the relative difference (or lack thereof) in expression levels of that gene under control and experimental conditions. The brightness of each spot corresponds to the total level of expression between the two experiments.
To interpret microarray data in detail, computer programs are required. However, general observations can be made by any informed observer. These mRNA are allowed to bind to the microarray chip at the same time. The ratio of red:green should reveal the relative levels of transcription under the two experimental conditions. So, a green spot shows that a specific gene was repressed under experimental conditions while a red spot reveals the induction of a gene. If a spot appears yellow, this means that the ratio was about 1:1 (a.k.a. no change in expression). For human viewing, this color scale is normally converted to a standard format (Figure 2).
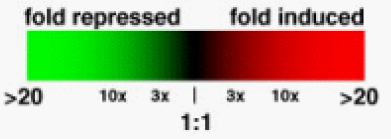
Figure 2. This figure, taken with permission from Genomics, Proteomics & Bioinformatics depicts a common color scale used by most researchers to depict the relative levels of translation in experimental (red) vs. control (green) conditions. Extreme repression and induction are represented by brighter green or red, respectively. As the difference in expression levels between the two populations gets smaller, the colors become dimmer, ultimately becoming black for a perfect 1:1 ratio (Campbell and Heyer, 2003).
Vas1 Expression
On my favorite yeast genes webpage, I reported that valyl-tRna synthetase, vas1, catalyzes the formation of valyl-tRNA. The valyl-tRNA goes on the deliver valine to the appropriate location in a developing polypeptide. Thus, vas1 plays a key role in yeast protein production. This same gene is alternately spliced to function in the cytoplasm and mitochondria of yeast cells(Chatton et al., 1987). The molecular function of this enzyme is to bind valine and an appropriate tRNA molecule, bringing them close and allowing them to bind. During this process, the enzyme cleaves two phosphate groups from a molecule of ATP, consuming 1 unit of this cellular energy molecule (Figure 2).
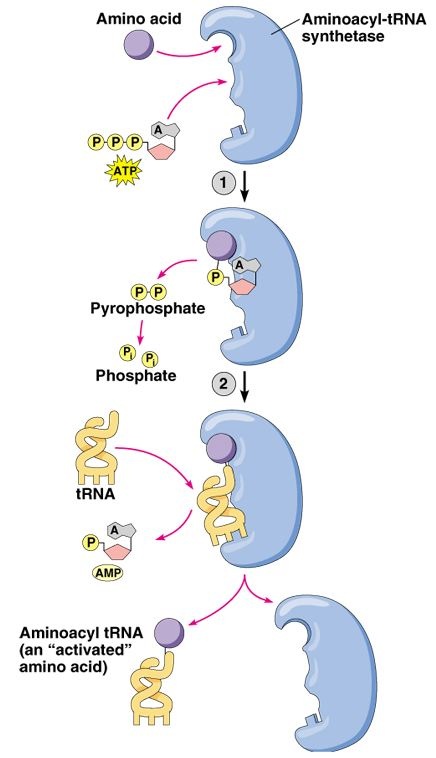
Figure 2 . Permission pending for this figure from Addison Wesley Longman. This figure depicts the general molecular function of an aminoacyl tRNA synthetase.
The Expression Connection website allows anyone with internet access to search for data from yeast microarray expression comparisons under different experimental conditions. In addition, users can easily search for other similarly expressed genes under these experimental conditions. I expect to find Vas1 clustered with other aminoacyl tRNA synthetases, since they are all required for protein production. Since Vas1 consumes a molecule of ATP through its molecular function, I expect to see its expression repressed under conditions where energy conservation is necessary.
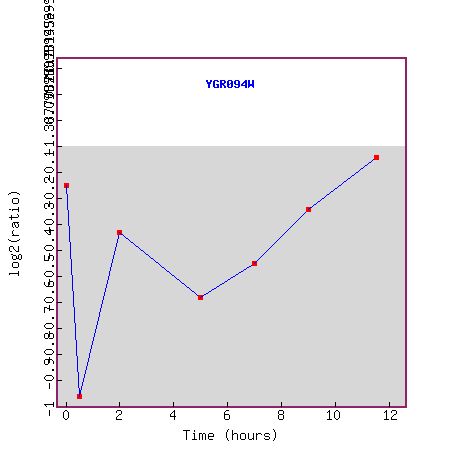
Figure 3. This graph, taken from the Expression Connection website, shows the expression of Vas1 (YGR094w) during several time points of sporulation. Although the Y-axis numbers unreadable, it shows the relative levels of expression over time. The line dividing the grey and white areas represents the control's level of vas1 expression. The further below that line a data point is, the more the gene is repressed under experimental conditions. The X-axis shows the time that has elapsed after sporulation. Vas1 is initially repressed within the first hour and slowly becomes less repressed as time elapses. Protein translation seems to be at its lowest at the very beginning of sporulation. Perhaps energy is conserved for other processes at this time. The figure below shows genes that exhibit similar expression patterns under these experimental conditions.

Figure 4. This Expression Connection image capture shows 20 genes that correlate closely in their expression patterns with vas1 during the sporulation stage of yeast. Chu and his colleagues hoped to identify genes that functioned in gametogenesis, especially those that were previously uncharacterized, during this experiment (1998). However, this experiment also provided much data on the entire yeast genome. While none of these similar genes are aminoacyl tRNA synthetases and many have unknown functions, most of the known genes code for proteins involved directly in translation. These results, while slightly unexpected, still support the notion that similarly expressed genes will have related functions.

Figure 5. This Expression Connection snapshot was taken from an search for similarly expressed genes to Vas1 under various conditions of environmental stress. Audrey Gash and a number of Stanford colleagues compared yeast expression under a large variety of environmental stresses (2000). Vas1 expression correlated closest to THS1, GRS1, MES1, and HTS1 among others. These genes are all aminoacyl tRNA synthetases. As I predicted, genes of very similar biological process correlate strongly, even under a wide variety of experimental conditions.
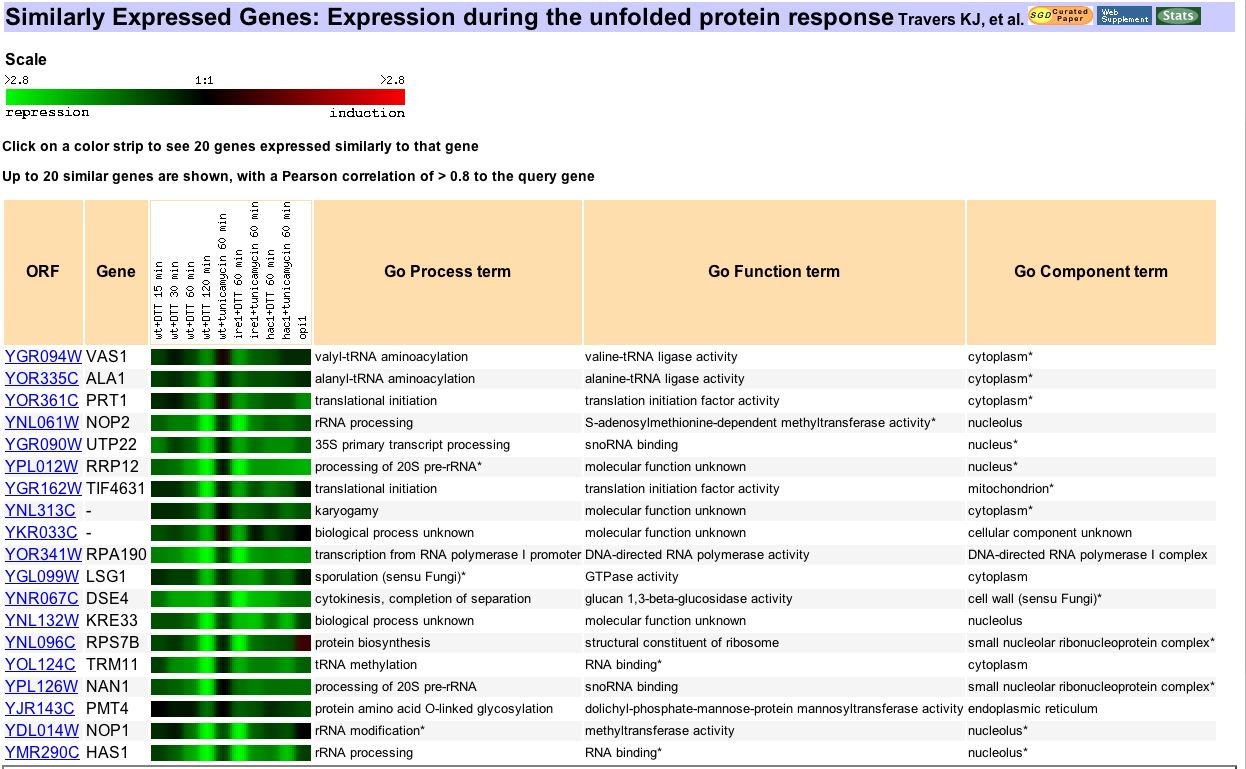
Figure 6. Another photo from Expression Connection, this image shows genes similarly expressed to vas1 during cellular response to protein folding errors. Kevin Travers and fellow researchers performed this microarray experiment to analyze the genome-wide results of an unfolded protein response (UPR) in the endoplasmic reticulum (2000). Once again, this experiment provides data about the entire yeast genome, including many genes that were not thought to have any role in this process. Vas 1 correlates closest to another aminoacyl tRNA synthetase, ALA1. Its expression also correlates to many different types of genes, most of which are somehow involved in protein translation. The UPR affects many genes involved in every stage of protein production. So, it is not surprising that Vas1 correlates with many genes of related biological processes in this experiment.
 Figure 7. Another snapshot from Expression Connection, this comes from a search of genes expressed similar to vas1 in yeast exhibiting different levels of polyploidy. In this experiment Galitski and partners used this experiment to identify the genes responsible for the effects observed in polyploid yeast (1999). The focus of this experiment was not genes involved in protein production. Vas1's expression profile correlates to genes of widely varying functions, from protein processing to shpingolipid metabolism. These results seem to contradict the assumption that genes of similar function will always correlate together. However, closer inspection reveals that almost every vas1 expression ratio from the data shows up as a dark color. This means that the degree of induction or repression was very slight if it occurred at all. Ratios this close can easily be skewed by chance variation. So this seemingly random assortment of correlated genes may be just that, random.
Figure 7. Another snapshot from Expression Connection, this comes from a search of genes expressed similar to vas1 in yeast exhibiting different levels of polyploidy. In this experiment Galitski and partners used this experiment to identify the genes responsible for the effects observed in polyploid yeast (1999). The focus of this experiment was not genes involved in protein production. Vas1's expression profile correlates to genes of widely varying functions, from protein processing to shpingolipid metabolism. These results seem to contradict the assumption that genes of similar function will always correlate together. However, closer inspection reveals that almost every vas1 expression ratio from the data shows up as a dark color. This means that the degree of induction or repression was very slight if it occurred at all. Ratios this close can easily be skewed by chance variation. So this seemingly random assortment of correlated genes may be just that, random.
Expression Connection Conclusions
Since the function of vas1 is well-characterized, it is a useful exercise to analyze the Expression Connection results of various searches. In this way, a known gene can serve to measure the reliability of this enormous database. In Figures 4, 5, and 6, the results supported my expectations. Under these conditions, vas1 correlates very similarly to genes of related molecular function or biological process. However, Figure 7 shows that careful analysis is required in interpreting results from this type of database. A search of Expression Connection might seem to produce startling, unprecedented results. However, this might be the consequence of random chance as much as it might be extremely meaningful. A careful researcher must understand all that the data reveals and not jump to conclusions. These lessons are especially important in analyzing the Expression Connection search results of an uncharacterized potential gene.
YGR093w Expression
On MFYG website, I explored the non-annotated ORF YGR093w. This predicted ORF has not been verified. While YGR093w has many characteristics of a gene's coding sequence, it does not code for a known protein. Since theYGR093w predicted protein shares conserved domains with Cwf-family genes (a family of genes involved in the spliceosome) and it is known to be expressed in the nucleus of yeast cells, I hypothesized that YGR093w took part in or interacted with the yeast spliceosome. On this webpage, I have analyzed the available database information on YGR093w's expression profile to test my hypothesis.

Figure 8. From Expression Connection, this figure is the result of a search for genes similarly expressed to YGR093w during sporulation. As noted earlier, this experiment was run by Dr. Chu and partners to identify genes that functioned in gametogenesis (1998). The data from this experiment shows that YGR093w was first repressed and then induced over the first 11.5h after sporulation began. The similarly expressed genes on this chart code for proteins involved in various types of biosynthesis, mRNA transport as well as mRNA splicing. While this may lend support to my hypothesis, I must avoid my bias by refusing to selectively ignore non-supportive data. The correlation to mRNA splicing gene LSM7 is encouraging, but the even stronger correlation to SNG1 is confusing. This data neither explicitly confirms nor disproves my hypothesis.
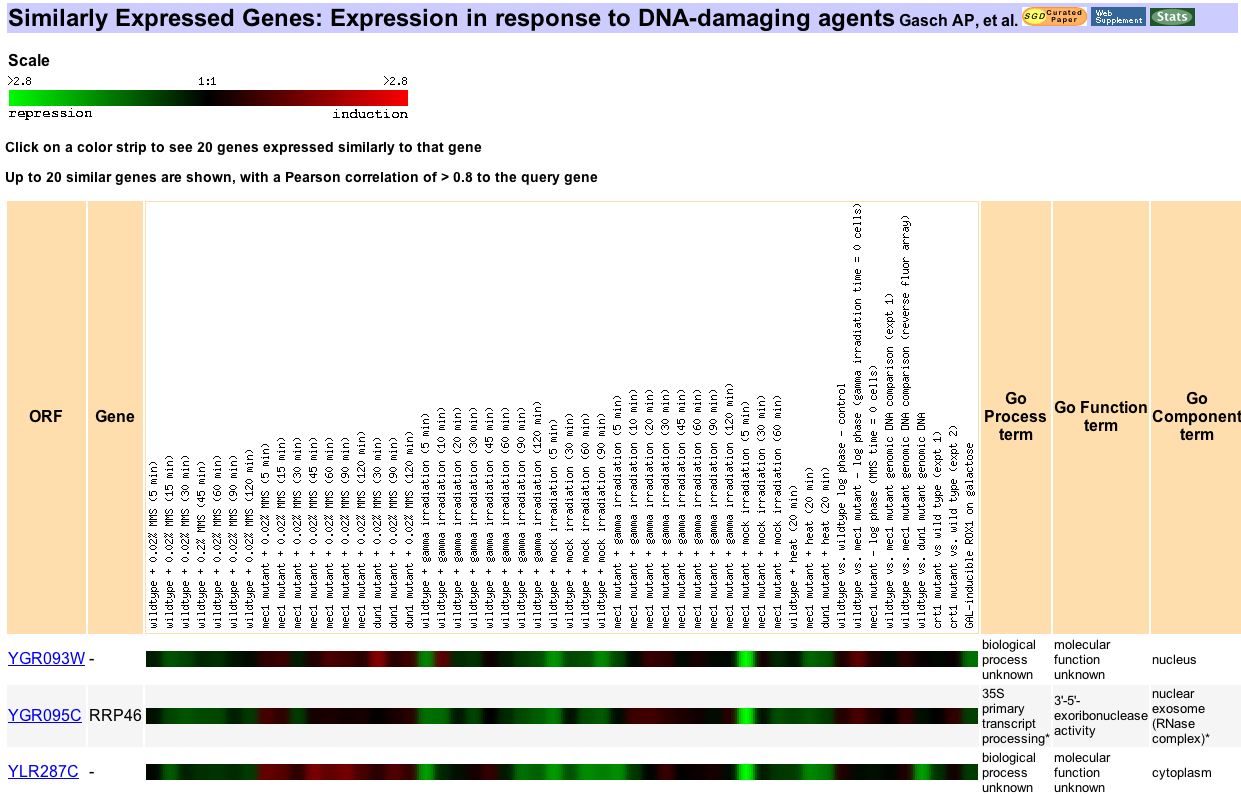
Figure 9. In this Expression Connection search for genes similarly expressed to YGR093w under treatment with DNA-damaging agents. Dr. Gasch and colleagues used this experiment to analyze the expression of MEC1 during DNA damage events as well as the effects of deleting this gene during such events (2001). YGR093w shows varying response to the experimental conditions. It is difficult to tell whether these expression changes are due to the loss of Mec1 or the DNA damaging events. Only two other genes correlate strongly enough to YGR093w to be shown and only one of them is annotated. However, RRP46 does indeed function in transcript processing. This result lends some further support to my hypothesis that YGR093w is involved somehow in RNA transcript alteration.

Figure 10. Using Expression Connection, these results come from a search of genes expressed similarly to YGR093w during unfolded protein response. Travers and company wanted to analyze the genomic results of this response pathway (2000). While they assumed that UPR would affect mainly ER-related genes, their results show that the cell's response extends to genes involved in many different biological processes. Earlier, I noted that Vas1 and many other genes involved in translation are repressed under this experimental condition. On this chart, it is evident that many genes involved in transcription are repressed as well. YGR093w is repressed similarly to several genes involved in rRNA processing. YGR093w's expression also correlates strongly to a few genes involved in translational initiation and one involved in protein biosynthesis. In light of these genes, the results from this search are not conclusive. However, evidence is mounting that YGR093w has a biological process related to transcription or transcript processing.

Figure 11. This final set of Expression Connection results comes from a search for genes expressed similarly to YGR093w in the presence of small-molecule inhibitors of rapamycin (SMIR). Jing Huang and associates at Harvard, Yale and UCLA collaborated to explore the effects of SMIRs on yeast expression. These experiments were designed to identify important genes in the targets of rapamycin pathway, thought to mainly code for nutrient signaling proteins (2004). The results are non-conclusive. YGR093w clusters with genes involved in DNA replication, mitochondrial organization and a lone gene, MSS116, involved in RNA splicing. However, some of the seemingly random correlations can be discounted. GPI4, RNR4, and MES1 are all encoded on the same chromosome. It would be erroneous to ignore the possibility that these proteins might cluster with YGR093w partially because of their nearby location.
YGR093w Expression Conclusions
On my MFYG website, I hypothesized that YGR093w had a biological process related to RNA splicing because it showed conserved domains and predicted protein similarity to many such genes. The results of my expression profile analysis neither confirm nor refute that hypothesis. It seems undeniable that YGR093w has some connection to transcriptional processing. Figures 8, 9 10, and 11 all show that YGR093w is expressed similarly under some experimental conditions to a number of genes with a biological process related to RNA splicing or at least transcription. However, the usefulness and limitations of this type of analysis have become apparent. An initial hypothesis can easily bias a researcher towards finding support for their educated guess. My hypothesis has actually been broadened instead of refined by these results. Now, I am more confident that YGR093w functions in some part of transcription, but I am no longer positive that it is directly involved in the functioning of the spliceosome.
References
Campbell A M, and Heyer L J,. 2003. Discovering Genomics, Proteomics, and Bioinformatics. Benjamin Cummings: San Francisco.
Chatton B, Walter P, Ebel J, Lacroute F, and Fasiolo F,. 1987. The Yeast Vas1 Gene Encodes Both Mitochondrial and Cytoplasmic Valyl-tRNA Synthetases. J Biol Chem. 261 (1): 52-57.
Chu S, Derisi J, Eisen M, Mulholland J, Botstein D, Brown P O, and Herskowitz I,. 1998. The transcriptional program of sporulation in budding yeast [abstract]. In Science 282 (5389): 699-705. SGD Curated Paper. <http://db.yeastgenome.org/cgi-bin/reference/reference.pl?dbid=S000055354>. Accessed 2005 October 19.
Galitski T, Saldanha A J, Styles C A, Lander E S, and Fink G R,. 1999. Ploidy regulation of gene expression [abstract]. In Science 285 (5425): 251-4. SGD Curated Paper. <http://db.yeastgenome.org/cgi-bin/reference/reference.pl?dbid=S000050073>. Accessed 2005 October 19.
Gasch A P, Huang M, Metzner S, Botstein S, Elledge S J, and
Huang J, Zhu H, Haggarty S J, Spring D R, Hwang H, Jin F, Snyder M, Schreiber S,. 2004. Finding new components of the target of rapamycin (TOR) signaling network through chemical genetics and proteome chips. PNAS 101 (47): 16594-16599. <http://www.pnas.org/cgi/content/full/101/47/16594>. Accessed 2005 October 19.
Travers K J, Pati C K, Wodicka L, Lockhart D J, Weissman J S, and Walter P,. 2000. Functional and Genomic Analyses Reveal an Essential Coordination between the Unfolded Protein Response and ER-Associated Degradation. Cell 101 (3): 249-258. <2F2000&_alid=326047313&_rdoc=1&_fmt=&_orig=search&_qd=1&_cdi=7051&_sort=d&view=c&_acct=C000058476&_version=1&_urlVersion=0&_userid=2665120&md5=d54204ddb00e250df6deca56dff43599>. Accessed 2005 October 19..
Links
Davidson College Biology Department
Microarray Methodology Flash Animation
© 2005 Department of Biology, Davidson College, Davidson, NC 28036
Please direct comments, criticisms and questions to andrysdale "at" davidson.edu