Abstract
Mutations in the XPA protein, a damaged DNA binding protein, lead to the autosomal recessive disorder called xeroderma pigmentosum (XP). The XPA protein is a component of the nucleotide excision repair (NER) process that specifically repairs pyrimidine dimers. The most common cause of pyrimidine dimers in the DNA structure is irradiation from solar UVB rays. Through the use of literature research and experimental research I was able to build a model of the NER process. The model of NER helped me make predictions about the effects of a mutation in the XPA protein. Within the NER process XPA is essential for the recruitment of the single stranded DNA binding protein RPA and the endonuclease ERCC1. In my study I specifically looked at 2 non-synonymous single nucleotide polymorphisms (SNP) that are located in an alpha helix secondary structure on exon 6. I predict that these SNPs change the structure of XPA in such a way that it can no longer recruit and bind to RPA. In the absence of RPA, ERCC1 cannot be recruited. Because these proteins are essential to the NER process, DNA cannot be repaired, leading to cancer at an early age. To treat this specific form of the disease I have propose the use of a liposomal lotion with functional copies of the XPA protein. Because of the delivery method and the target location of this therapy, XPA proteins will be able to easily diffuse directly into the epithelial cells and take part in the now functional NER process.
Background
How do UV rays Damage DNA?
The sun emits ultraviolet radiation that travels through space to the earth. This radiation can be very harmful to the cells of organisms that have direct contact with the sun, skin. The ozone layer can block the shorts, and most dangerous, of UV rays called UVC rays. Unfortunately, the ozone cannot block everything. Longer less energetic wavelengths can past though the ozone layer and reach the organisms on earth. The wavelengths that are just long enough that they can get past the ozone, but not short enough that they are not harmful are UVB rays [Goodsell 2007].
When the sun hits your skin it is inevitable that UVB rays pass through your cell walls and interact with your DNA. The UVB rays interact with the pyrimidine bases of DNA and break bonds causing single stranded lesions within a double stranded segment of DNA. If this happens to two bases that are next to each other they will covalently bond forming a tight little ring within the DNA structure. If there is only a single pyrimidine bond broken it will form a single bond with two carbon atoms. This lesion is called a pyrimidine dimer [Goodsell 2007]. Pyrimidine dimers are on a single strand of DNA within a double stranded DNA complex.
Pyrimidine dimer reactions are very common and occur 50 to100 times within one cell during one second of sun exposure [Goodsell 2007]. Fortunately, for most of us, the pyrimidine dimer is corrected seconds after it forms by the nucleotide excision repair (NER) process. The proteins involved in NER work together to find lesions in the double stranded DNA caused by pyrimidine dimers on one of the strands. The proteins bind to the single stranded region of the lesion where the pyrimidine dimer is located. They then unwind the double stranded DNA around the pyrimidine dimer and cut out a section around the dimer that is usually about 30 bases in length. After the pyrimidine dimer is removed the normal DNA replication machinery can fill in the gap. For humans, NER is the only defense that we have against UV damage [Goodsell 2007]. Therefore, without NER our cells have no defiance against UV damage. Most of the time pyrimidine dimers are not harmful, but with 50 to 100 happening every second that you are in the sun the probability of one being in a location that happens to cause cancer increases greatly if the lesions are not repaired. In patients with XP this is exactly what happens. There NER is either not working at all or its function is greatly diminished. Therefore, exposure to the sun is very harmful to XP patients, even if it is just for a few seconds.
The Disease
The XPA gene is part of the highly conserved nucleotide excision repair (NER) pathway which repairs damaged DNA. When XPA is faulty the NER cannot repair damaged DNA causing a rare autosomal recessive condition called xeroderma pigmentosum (XP) [Riedl et al. 2003]. Affected persons usually will develop acute sensitivity to the sun between 1 and 2 years of age. XP is first manifested in prolonged redness and blistering due to sun exposure [Lin et al. 2004]. Sun damage will accumulate due to inevitable sun exposure. This sun damage leads to the development of multiple skin cancers (including basal cell carcinoma, squamous cell carsinoma, and melonoma) at a very young age [Lin et al. 2004].
References
1. Goodsell DS. The molecular perspective: Ultraviolet light and pyrimidine dimers. Stem Cells 2001;19(4):348-9.
2. Lin P, M.D., and English JCIII, M.D. Topical treatment of xeroderma pigmentosum Continuing Medical Education 2004;29(8):512-514.
3. Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 2003 Oct 1;22(19):5293-303.
XPA
XPA “protein binds to damaged DNA as a damage-recognizing protein” that is located in the nucleolus of human cells [Miyamoto et al. 1991]. Specifically XPA is part of the nucleotide excision repair (NER) process. The NER specifically binds to pyrimidine dimers that are caused by UVB rays from the sun (as described in the introduction). The gene that encodes XPA is located on chromosome 9q22 and consists of 6 exons. Exon 1 is involved in cellular localization, but is not needed. Exon 2 encodes the glutamate acid cluster, and exon 3 encodes the zinc figure structure both of which are critical for repair function. The long region from Exons 2 to exon 6 is essential for DNA repair function [Miyamoto et al. 1991].
XPA GO terms [Ensemble Genome Browser]
Biological functions
binds damaged DNA binding, zinc ions,
metal ions, nucleotides, and proteins
Molecular process
nucleotide-excision repair
Cellular component
Nucleus
Conserved Domains [NCBI CDD]
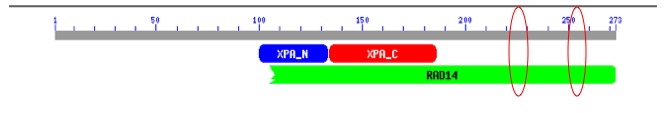
XPA has three conserved domains, XPA_C, XPA_N, and RAD14. XPA_C is the C-terminus of XPA. XPA_N is the conserved domain for the N-terminus of XPA. RAD14 is a DNA binding domain (Figure 1).
Clustal Analysis [EBI Tools: Clustal W]
Conservation among species for the XPA gene is particularly high in mammals. A ClustalW analysis comparing XPA to a variety if species showed the following percent similarity between the species:
| Species | % Similiarity |
| Mus musculus (Mouse) | 84 |
| Pan troglodytes (Chimp) | 99 |
| Drosophilinae (Fruit Fly) | 45 |
| Caenorhabditis elegans ( Worm) | 35 |
| Saccharomyces cerevisiae (Yeast) | 25 |
| Felis catus (Cat) | 93 |
| Canis familiaris (Dog) | 92 |
As you can see it seems that the XPA gene is most highly conserved among mammals. This is not surprising because in mammals the NER is the only defense against UV damage. Another factor to consider is that most mammals evolved from nocturnal ancestors, and therefore only needed one way of repairing damage from UV rays.
Motifs [JASPAR]
Because XPA interact with other proteins I was wondering what kinds of motifs it has in its coding region. Therefore I performed a JASPAR database search of the coding region of XPA with a threshold of 80%, and found that the XPA protein has the following motifs:
- C2H2 zinc finger binding region called ZNF354. Indicated that this zinc finger motif is found on the leading strand at base pairs 571 to 577. This comparison had a relative score of 1.000, and a score of 8.916.
- ETS binding region called SPI1 with a score of 8.711, a relative score of 1.0 and is found on the leading strain from base pair 153 to 158.
- bHLH-ZIP binding region called MAX with a score of 9.603, a relative score of 0.894 and found on the lagging strand from base pair 1308 to 1317.
I also looked at the promoter region to see what kinds of transcription factors were transcribing XPA and I found:
- C2H2 zinc finger binding motif called YY1 with a score of 7.027, a relative score of 0.931, and is found on the lagging strand 233 base pairs up stream of the XPA gene.
- A TATA Box with score 7.444, relative score of 0.835103, and is found base pairs up stream of the gene.
- AP2 binding region that is named TFAP2A. It has a score of 8.056, relative score of 0.924 and is found on the leading strand 578 base pairs upstream of XPA.
XPA SNPs [Ensemble Genome Browser]
XPA has a total of 23 single nucleotide polymorphism (SNP).
| Classification | Sub Number | Number |
| Downstream | 2 | |
| 3 Prime UTR | 4 | |
| Non Synonymous Coding | 5 | |
Multiple base insertion |
1 (4 nt.) |
|
Multiple base deletion |
1 (3 nt.) |
|
| Synonymous Coding | 2 | |
| Franshift Coding (insertion) | 1 | |
| Intronic | 7 | |
| 5 Prime UTR | 1 | |
| Upstream | 1 |
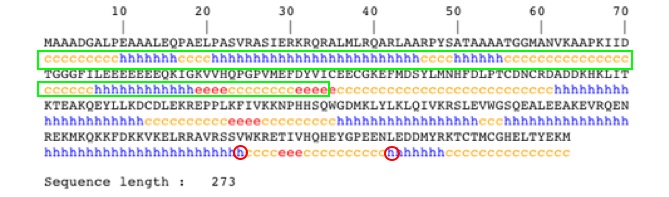
I have chosen to look at two non-synonymous SNPs because of their location within the XPA protein (Figure 1). The two SNPs are located at amino acids 234 and 252. The SNP at amino acid 234 changes a Valine to a Leucine causing an increase in the hydrophobic index from 4.2 to 3.8. The SNP at amino acid 252 conversely changes a Leucine to a Valine which causes an increase in the hydrophobic index from 3.8 to 4.2 [Amino Acid Properties]. With in the protein as a whole they are C-terminally located on the 6th exon. In addition, these SNPs are located in a highly conserved DNA binding region in a alpha helix secondary structures.
A)
|
B)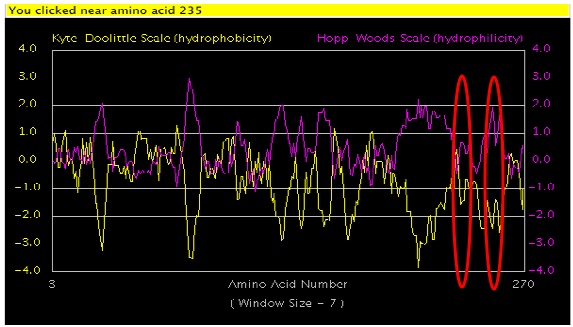 |
C) |
|
Figure 1: Click figures to enlarge. SNP locations are at amino acids 234 and 252. In parts A through B SNPs are circled in red. A) Predicted 2-D structure [Predator]. The blue “h” represents alpha helix structure, yellow “c” represents random coils, and red “e” represents extended strains. SNP mutations are found in alpha helix regions. Green box shows area was low conservation. B) Kyte doolittle (testing hydrophobicity) test is in yellow, and Hopp Woods (testing for hydrophilcity) test is in purple. Window size is 7. Shows that the protein XPA is neither strongly hydrophobic nor hydrophilic. Proves that XPA is not a membrane or trans-membrane protein.[Hydropathicity Plots] |
C) Conserved domains of XPA. Red region of XPA_C is the C-terminus of XPA. XPA_N is the conserved domain for the N-terminus of XPA. RAD14 is a DNA binding domain. XPA_C is the most highly conserved (e-value=7e-21), RAD14 is the next most highly conserved (e-value=9e-16), and XPA_N is the least conserved (e-value=1e-10) [NCBI CDD]. |
Conclusion
In a study done by Miyamoto et al. in 1992 they performed specific mutations on XPA and observed the effects of these mutations. One of their cell lines contains had a mutation that removed amino acids 226 through 273, and consequently included my SNPs of interest. They found that the deletion of amino acid 226 through 273 did not effect the cellular localization of XPA, but did effect the level at which DNA was repair in response to UV damage. The deletion of 226 to 273 only had marginal levels of DNA repair. This suggests that a non-synonymous SNP in this region would greatly decrease the level at which DNA is repaired.
The means by which XPA repairs DNA is by working with other proteins in the NER process. During this process XPA probably binds to other proteins to form the complex in the NER process that repairs DNA. Because the SNPs that I am looking at are not located in the DNA binding zinc finger structure encoded for in exon 3, I do not think that they disrupt the ability of XPA to bind to DNA. Instead I hypothesis that these two SNPs in question affect the ability of XPA to bind to other essential proteins and form the NER complex. Because the complex is not formed correctly it does not function properly. Therefore, DNA damaged by UVB rays does not get repaired.
To test this hypothesis I would be specifically looking at whether or not the NER complex was being formed when the two SNPs in question are present in the XPA protein. To answer this I would use the process of immunoprecipitation. Immunopercipitation uses an antibody that binds to the the protein in question, in this case it is XPA. I will then be able to pull out XPA and the proteins that it is bound to. In order to test my hypothesis I will need to have one experiment with a wild type version of XPA and one that will have to be induced to have the specific SNPs that I am interested in. By comparing the results from the two experiments I would be able to tell if the mutations in XPA had an effect on the formation of the NER complex in relation to XPA.
What if the SNPs cause nothing to bind to XPA?
I know that XPA is essential to the formation of a critical structure in the NER process [Riedl et al. 2003]. Therefore if nothing bond to XPA I would know that DNA was not being repaired because XPA would not be part of the necessary complex.
References
1. Amino Acid Properties [http://www.mcb.ucdavis.edu/courses/bis102/AAProp.html]
2. EBI Tools: Clustal W [http://www.ebi.ac.uk/Tools/clustalw/index.html]
3. Ensembl Genome Browser [http://www.ensembl.org/index.html]
4. Hydropathicity Plots [http://www.vivo.colostate.edu/molkit/hydropathy/index.html]
5. The JASPAR Database [http://jaspar.genereg.net/]
6. NCBI Conserved Domain Database (CDD) [http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml]
7. Predator [http://bioweb.pasteur.fr/seqanal/interfaces/predator-simple.html]
8.Miyamoto I, Miura N, Niwa H, Miyazaki J, Tanaka K. Mutational analysis of the structure and function of the xeroderma pigmentosum group A complementing protein. identification of essential domains for nuclear localization and DNA excision repair. J Biol Chem 1992 Jun 15;267(17):12182-7.
9. Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 2003 Oct 1;22(19):5293-303.
The next step in my research was to look at the expression of XPA in microarray experiments from yeast. The yeast otholog of XPA is RAD14.
Why Yeast?
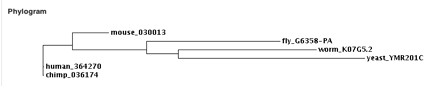
What makes us think that there can be any comparison between yeast and human genes? Yeast and humans are very far apart evolutionarily, but all living organisms have similarities and needs for the same basic processes. Repairing damaged DNA is one of those processes. The nucleotide excision repair (NER) process is highly conserved [Thorel et al., 2004], and specifically the most important region of XPA for DNA repair exons 2 through 6 are highly conserved from drosophila to humans [Miyamoto et al. 1991].
Figure 1: The Phylogram was obtained from a ClustalW analysis comparing the amino acid sequence of the XPA orthologs of Mus musculus (Mouse), Pan troglodytes (Chimp), Drosophilinae (Fruit Fly), Caenorhabditis elegans ( Worm), and Saccharomyces cerevisiae (Yeast) to the human XPA protein sequence. XPA conserved domains XPA_C is the most highly conserved (e-value=7e-21), RAD14 is the next most highly conserved (e-value=9e-16), and XPA_N is the least conserved (e-value=1e-10). RAD14 conserved domain RAD14 is the most high conserved (e-value=9e-92). XPA_C is the other conserved domain within RAD14 which is not as highly conserved as RAD14 (e-value=3e-07) [NCBI Conserved Domains].
ClustalW Comparison of RAD14 and XPA
When comparing the human to the yeast of the whole coding region of the XPA protein there was a 25% similarity. As you can see in the phylogram in figure 1, the yeast version of XPA (RAD14) and the human XPA are far apart evolutionarily. This dissimilarity between yeast and humans could be made larger by the fact that the RAD14 protein is 371 amino acids long while the human XPA protein is 273 amino acids long.
Comparison of RAD14 and XPA GO Terms [Ensemble Genome Browser]
Biological functions
Both: damaged DNA binding and zinc ion binding proteins
XPA Only: also binds metal ions, nucleotides, and protein
Molecular process
Both: nucleotide-excision repair
RAD14 Only: also is a DNA damage recognition protein
Cellular component
Both: Nucleus
The GO terms are very similar making it evident that the two proteins RAD14 and XPA play similar roles within their respective organism.
Microarray Research
Using data collected by the Hoops Lab at Washington University I analyzed the expression of RAD14 by comparing three experiments
- Wild type stationary phase growth vs. log phase growth (control)
- msn2/msn4 deletion mutant stationary phase vs. wild type log phase growth
- msn2/msn4 deletion mutant in stationary phase vs. msn2/msn4 deletion mutant log phase growth
Msn2/msn4 is a transcriptional activator. It is activated in response to stress, which results in the translocation of msn2/msn4 from the3 cytoplasm to the nucleus where it binds DNA at stress response elements of responsive genes [SGD].
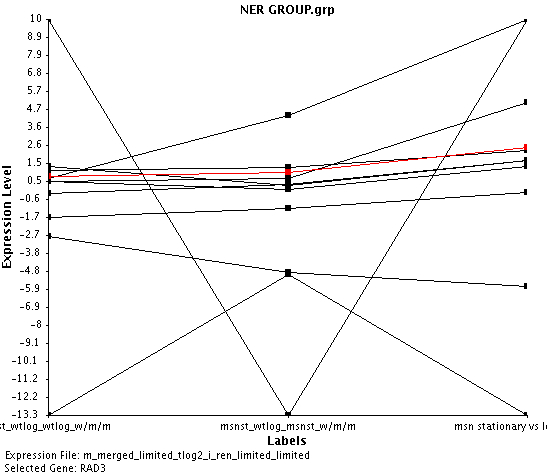
I found that RAD14 was being repressed in the conditions, and that it was being more pressed as the msn2/msn4 transcription factor was being deleted. This tells me that msn2/msn4 must transcribe RAD14.
In these conditions I found that a few genes were co regulated with RAD14 with a dissimilarity coefficient of 0.0005, but none of these genes were involved in the repair of damaged DNA. Then I looked at just the genes that had a biological process of DNA damage, and surprisingly did not find any of the genes that were part of the nucleotide excision repair (NER) process (Figure 2). What does this mean? It could mean a few things. First, it could mean that the proteins involved in the NER process are not being expressed in equal amount, even when they form heterodimers. Secondly, that for some reason or another the level of mRNA does not directly correlate to the level of proteins. Thirdly, it could mean that the NER process is not active.
If the third option were true, what would that mean?
We know that the NER process is activated in direct response to the formation of pyrimidine dimers, which are most often formed by UVB irradiation [Goodsell 2007]. This experiment did not have the stress of DNA damage caused by UVB rays. Therefore, if UVB irradiation is the only kind of stress that turns on the NER process we would not expect to see much co regulation of the genes involved in the NER process when this stress is absent. Furthermore, it follows that only when the NER process is active would we see co regulation of the genes involved. Like wise, a lack of co regulation of these genes would suggest that the NER process is not active. To test if this line of logic is true we would need to see if the genes involved in the NER process were co regulated in a condition that we knew induced activity of the NER process, like UVB irradiation. (Note: The lack of co regulation even under a condition such as UVB damage does not absolutely mean that the logic here is wrong. Yeast and humans are very different and the NER process may not be the same in yeast as it is in humans) If the logic is correct, and there is co regulation of these genes under UVB irradiation, we would be able to test the specificity of the NER process to different kinds of DNA damage.
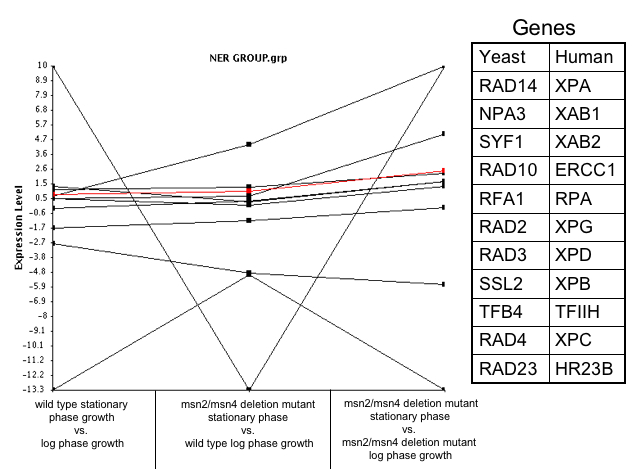
Figure 2: Plot shows the expression ratios of genes that are involved in the NER process under the three Hoopes Lab experiments. Table in the figure gives the human and yeast genes involved in the NER process.
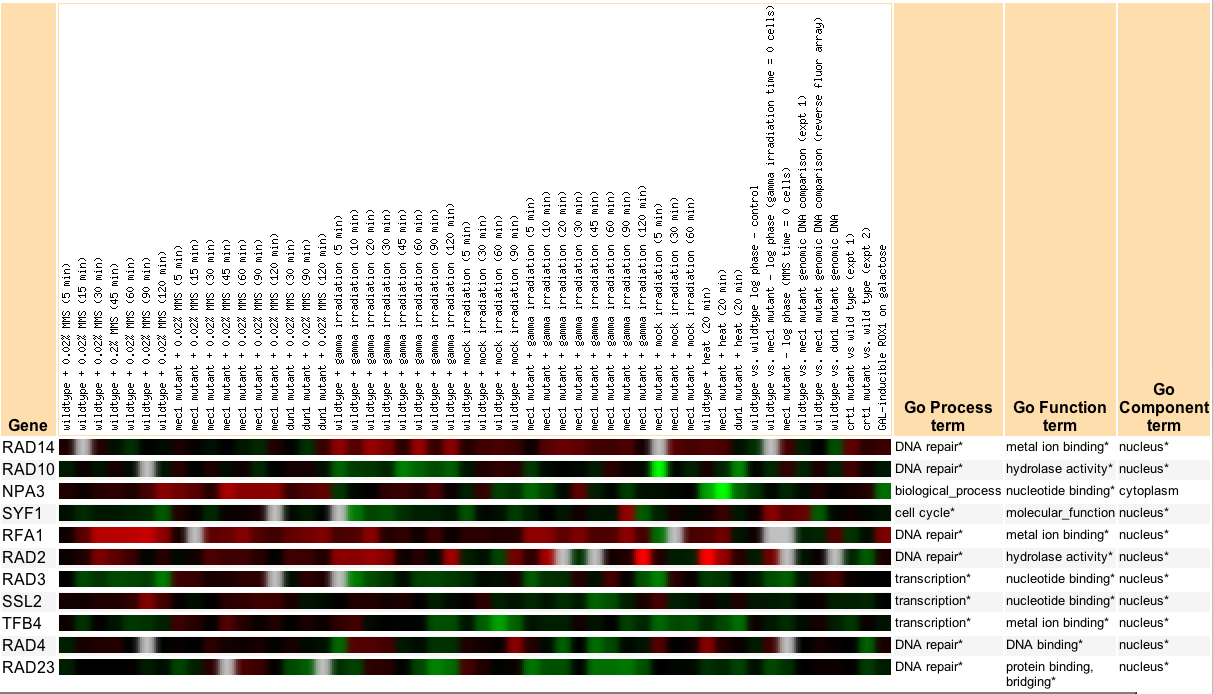
DNA Damaging Reagents Experiment
I looked at the expression of the proteins involved in NER in an experiment conducted by Gasch et al. where yeast was subjected to different DNA damaging conditions (Figure 3). There was no co regulation between the genes involved in the NER process in any of the conditions of this experiment. Out of the conditions that were tested I most expected the gamma irradiation damage to induce the NER process because it is the most like UV damage, but this was not the case. To try and explain this we will have to look into the differences between gamma radiation and ultraviolet radiation.
.
(Click to Enlarge)
Figure 3: Data obtained from SGD Expression Connection search engine. Experiment was with different DNA damaging reagents. It was performed by Gasch et al. in 2001.
Gamma vs. Ultraviolet
The genes that are involved in the nucleotide excursion repair (NER) process identify pyrimidine dimers on the DNA and cut them out. A study by Freeman et al., in 1989 looked at the wavelength spectrum at which pyrimidine dimers were formed. They found that the dimers are formed best by wavelengths of 300nm, and that if the wavelength is larger or smaller then 300nm it does not form dimers at as high of a frequency. Therefore, this means that when DNA is being damaged by radiation that has a wavelength that is larger or smaller then 300nm the genes involved in the NER would not be as active because there would not be as many pyrimidine dimers for them the to cut out. UVB has a wavelength between 290nm to 320nm, conversely gamma rays have a wavelength between 0.03nm to 0.003nm. With this in mind we can come to the conclusion that the NER process would be most active under UVB irradiation, and less active under gamma irradiation. Now that we have figured out the differences between gamma rays and UVB rays the results from the DNA damaging reagent experiment by Gasch et al.are not surprising.
Conclusion
My results suggests that the NER process is activated only in the presents of a very specific kind of DNA damage, pyrimidine dimers, that comes from a very specific source, UVB rays.
To test this hypothesis we would need to see what conditions the genes involved in the NER process are co regulated in. It would be informative to see what kinds of sources, including chemicals, induced the NER process. This kind of information would be useful because it would give us information on the cancer causing potential of a given source. To do this you could simply treat a set of cells with the source in question. You could then run a microarray that looked at the ratio between untreated cells and treated cells. If there was a significant up regulation of the genes involved in the NER process you could conclude that the source that was being used to treat the cell had cancer causing potential.
TRIPLES Experiment
In the TRIPLES database I found mTn insertions available for for yeast genes of the human orthologs XAB2, XPG, XPD, XPB, TFIIH, and HR23B. The mTn insertion basically means that the gene you are inserting the mTn into will not be transcribed. This kind of information does not help me answer relevant questions.
Future Experiment
Question: What DNA damaging reagents activate the NER process?
Experimental Design:
- Cy3 = wild type yeast cells that are not treated with any thing, and are grown in normal lab conditions
- Cy5= wild type yeast cells that are grown under a DNA damaging reagent
Control: For a base line reference I would use the comparison of yeast cells grown in normal lab conditions compared to those grown in UVB irradiation.
Conclusions that could be drawn: This would give me information on which DNA damaging reagents caused pyrimidine dimer lesions in DNA. This would also potentially be a way to tell how cancer causing a substance/treatment is when it touches human skin.
References
1. EBI Tools: Clustal W [http://www.ebi.ac.uk/Tools/clustalw/index.html]
2. Ensembl Genome Browser [http://www.ensembl.org/index.html]
3. Expression Connection [ http://db.yeastgenome.org/cgi-bin/expression/expressionConnection.pl]
4. Freeman SE, Hacham H, Gange RW, Maytum DJ, Sutherland JC, Sutherland BM. Wavelength dependence of pyrimidine dimer formation in DNA of human skin irradiated in situ with ultraviolet light. Proc Natl Acad Sci U S A 1989 Jul;86(14):5605-9.
5. Goodsell DS. The molecular perspective: Ultraviolet light and pyrimidine dimers. Stem Cells 2001;19(4):348-9.
6. Gasch AP, Huang M, Metzner S, Botstein D, Elledge SJ, Brown PO (2001) Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol Biol Cell 12(10):2987-3003
7. The JASPAR Database [http://jaspar.genereg.net/]
8. Miyamoto I, Miura N, Niwa H, Miyazaki J, Tanaka K. Mutational analysis of the structure and function of the xeroderma pigmentosum group A complementing protein. identification of essential domains for nuclear localization and DNA excision repair. J Biol Chem 1992 Jun 15;267(17):12182-7.
9. NCBI Conserved Domain Database (CDD) [http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml]
10. Saccharomyces Genome Database (SGD) [http://www.yeastgenome.org/]
11. Thorel F, Constantinou A, Dunand-Sauthier I, Nouspikel T, Lalle P, Raams A, Jaspers NG, Vermeulen W, Shivji MK, Wood RD, Clarkson SG. Definition of a short region of XPG necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol Cell Biol 2004 Dec;24(24):10670-80
12. TRIPLES Database [http://ygac.med.yale.edu/triples/default.htm]
XPA Gene Circuit
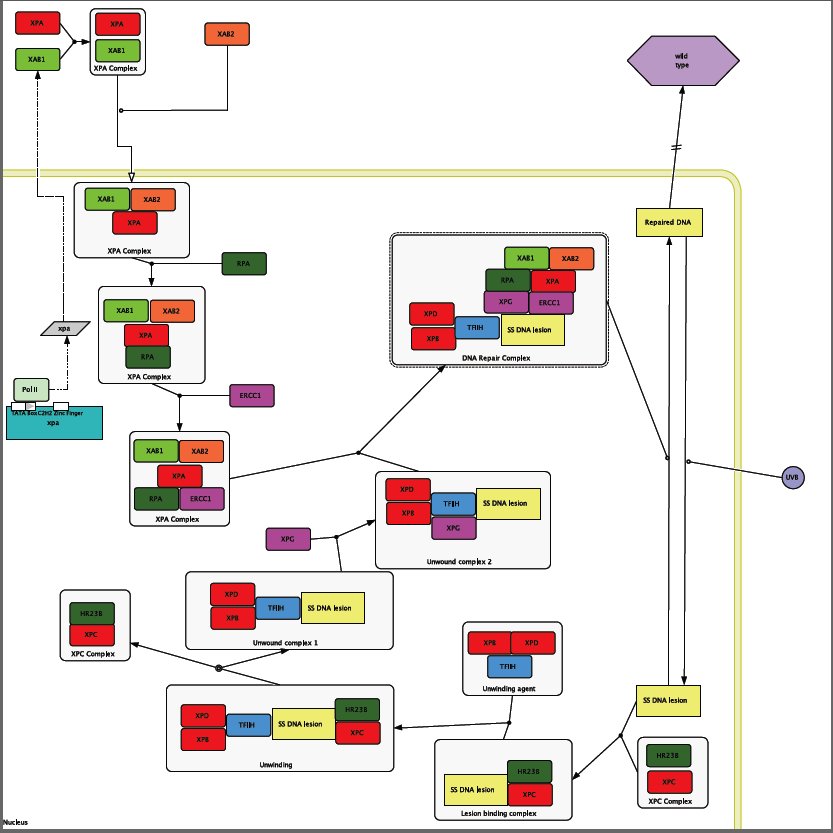
XPA is part of a process called the nucleotide excursion repair (NER) process. NERs main function is to repair DNA that has been damaged by UVB rays from the sun. In order to find the genes that are interacting with XPA in this circuit I performed a search on the human interactome map site [HiMAP].

(Click to Enlarge)
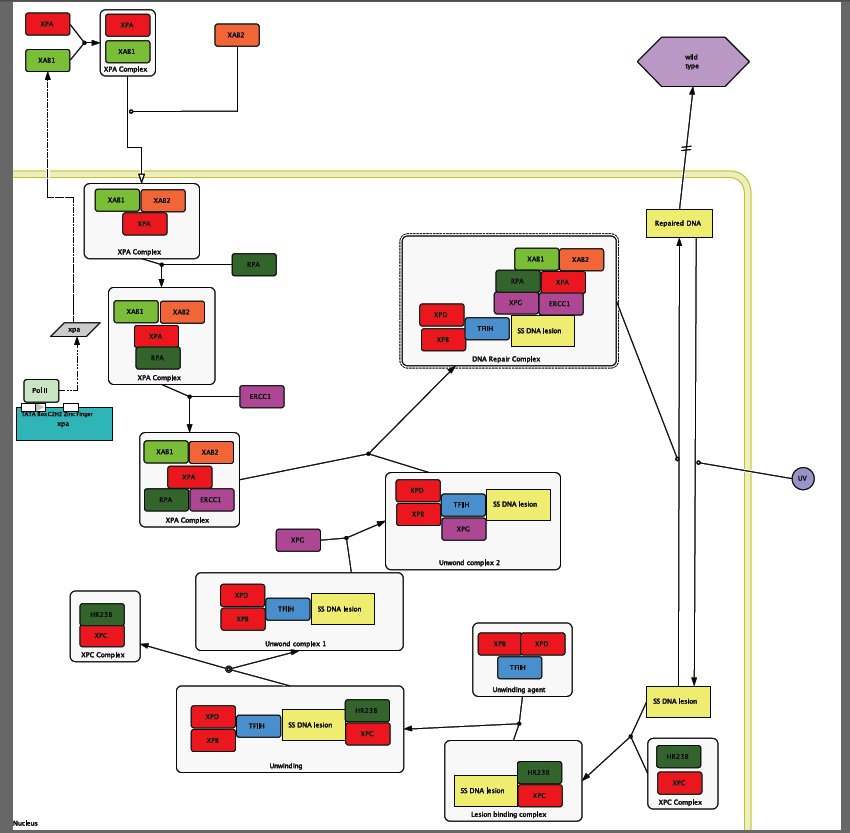
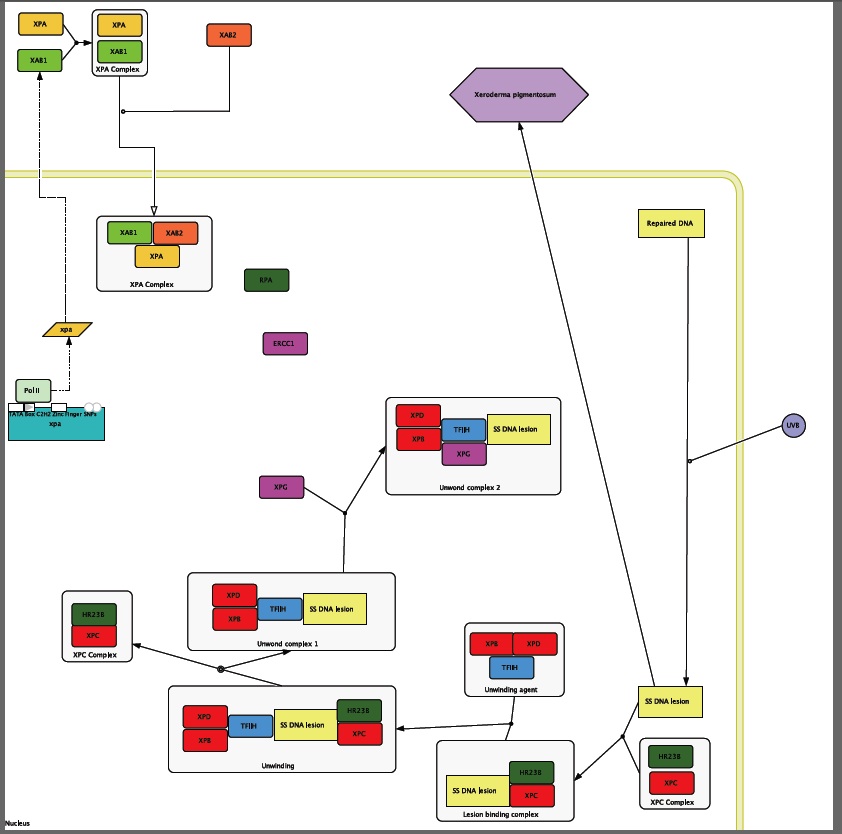
Figure1: Proteins that are circled are ones that I used to form my circuit diagram. Proteins that have highlighted stems are experimentally proven to have interactions with XPA.
I used the interactome in Figure 1 to start the formation of my circuit diagram. In addition to the information that I found in HiMAP I also used the information found by Riedl et al. when they performed an experiment in 2003 that explored the proteins involved in the NER process.
Strolling Through the NER process
XPA is transcribed and then transported out of the nucleus so that it can be translated and then folded. When it is out side of the nucleus it runs into the XPA binding protein called XAB1 that is an ATPase protein that is found in the cytoplasm [NCBI CDD]. Then this new heterodimer runs into another XPA binding protein, XAB2, which is also a trans membrane transporter protein. After inside the cell the XPA complex recruits the single stranded DNA-binding protein called RPA. Now that RPA has bond to the XPA complex it can now recruit the enonuclease ERCC1. This complex cannot form without ATP [Riedl et al. 2003], which is most likely provided by XAB1.
Unwound complex 1 Formation
UVB rays hit the skin during sun exposure and travel into the cell where they cause the formation of a prymimidine dimer that is recognized by an XPC complex that is comprised of single strand DNA bind protein HR23B and the DNA binding protein XPC forming the lesion binding complex (Kohn). This process does not need ATP to happen [Riedl et al. 2003]. Next the Lesion binding complex runs into the unwinding agent complex that is comprised of the DNA binding proteins XPB and XPD and the DNA repair protein TFIIH. These two complexes interact to unwind the DNA around the lesion [Thorel et al. 2004]. Then the presence of the XPA complex increases the release of the XPC complex from the now unwound DNA lesion [Riedl et al. 2003]. Then the unwound complex 1 recruits the endonuclease XPG to form the unwound complex 2 [Thorel et al. 2004].
The XPA complex and the unwound complex 2 come together to form the DNA repair complex. When the DNA Repair complex is together XPG can cut the lesion at the 3 prime end and ERCC1 can cut the lesion at the 5 prime end. The piece if DNA that is cut out is about 30 base pairs in length. After the DNA Repair Complex cuts out the mutation the cells normal DNA replication machinery can come in and fill in the gap [Goodsell 2007]. This results in repaired DNA. (Note: see introduction for more information)
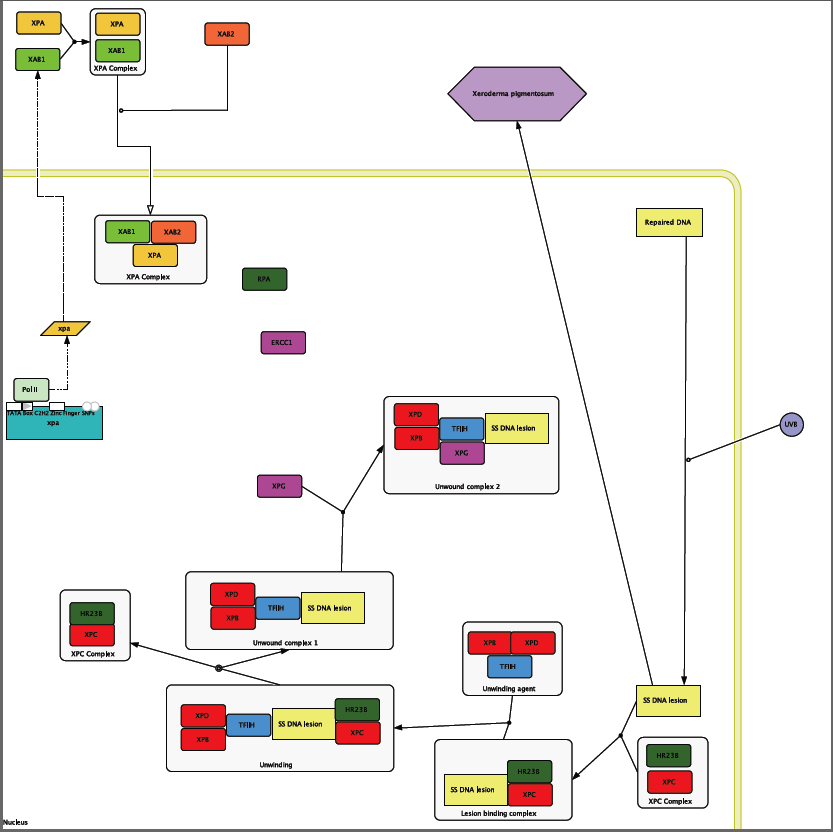
The Cause of XP
The two SNPs that are identified in the section on the XPA protein are transcribed into the mRNA, and then translated into the protein out side of the cell. XPA is able to bind to both XBA1 and 2, but because of the location of the SNPs XPA is not able to bind to RPA, and therefore the XPA complex with RPA cannot form. RPA is needed to recruit ERCC1 and form the Unwound complex 2 [Riedl et al. 2003]. Because XPA cannot bind to RPA the DNA Repair Complex is not formed and the pyrimidine dimer that is formed from UVB damage is not removed. Therefore, the mutations within the DNA caused by UVB rays accumulate over time, eventually causing skin cancer at a young age.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Assumptions Made in the Model
XAB1 is the protein that gives energy to the NER process
Testing: to test if XAB1 is giving energy to the NER process I would first need to test that XAB1 is part of the DNA repairing complex. To do this I would use immune, where I would manufacture a antibody that bond to XAB1. I would then place the cell under UVB irradiation so that I would know that the NER process was active. As a point of comparison I could also pull down XPA using the same method but with an antibody that was for XPA. By comparing these two products I will be able to see if XAB1 is part of the DNA repair complex. If I did find that XAB1 is part of the DNA repair complex I would then need to test and see if it dephosphorylates when part of the complex. To do this I would use a technique that uses microwells to test what proteins are dephosphorylating a substrate. The technique involves adhering a radioactively labeled substrate that can be dephosphorylated to the bottom of a well. The you take a protein that you are interested in, in this case XAB1, and put it into the well. the level of radioactivity that is observed will directly correlate to the amount of dephosphorylation that is occurring.
XAB1 and XAB2 are part of the NER process
Testing: To test this I would use immunopercipitation with anti bodies for XAB1, XAB2, and XPA. I would perform three separate experiments, and then compare the results. If the three proteins are all included in the final DNA repair complex, I should get the same results for all three experiments.
Treatments
Existing Treatments for the Phenotype
- Dermatome shaving or dermabrasion [Lin and English 2004]
- Oral isotretinoin [Kraemer et al. 1988]
None of the existing treatments help with the real biological problem, they just treat the effects of XP.
Potential in What Other Organisms
Other organisms that do not descend from nocturnal animals that did not need as much defense against UV rays, such as humans, have a back up plan with the NER is not working.
- The T4 bacteriophage has an endonuclease, called V, which simply cuts out the damaged bases. This endonuclease is very unique. It does not seem to specifically recognize the pyrimidine dimers as the NER does, but rather it recognizes the weakening of the DNA helix by the dimer. V binds to the weakened area of the DNA and kinks the DNA at the site of the dimer. It then flips one of the adenine bases away from the dimer lesion and into a pocket that is in the protein [Goodsell 2007]. A liposome lotion has been created that contains V. The lotion has been shown to increase the removal of pyrimidine dimers when applied to affected areas after sun exposure, and it had no adverse effects [Lin and English 2004].
- Fish, reptiles, marsupials, and plants use an enzyme called photolyase. The enzyme binds to pyrimidine dimers and removes them from the DNA when exposed to light with wavelengths between 300 and 500nm. When photolyase was applied to the skin in the form of a liposomal lotion and combined with the light treatment there was an increase in DNA repair [Lin and English 2004].
Proposed Treatment
The treatments that were proposed above are innovative and use mechanisms that other species have evolved to repair DNA. They apply to a broad range of phenotypes of XP. The therapy that I am proposing would be specific to XPA, but could be applied to other phenotypes of XP that originate with just protein. Because the disease affects the skin the easiest way to deliver a therapy would be by using liposomes that you can fill with what ever you want. This way the liposome and its cargo can simply be applied to the skins surface, in the form of a lotion, and they will just defuse through the lipid membrane of the skin cells. The specific mutation that I have been looking at suggests that XPA has a mutation that causes it to not bind properly. Therefore, if we just stick a healthy version of the XPA protein into the liposome it can be easily delivered into the epithelial cells that are experiencing damage from UVB rays. The liposomal lotion would need to be applied regularly because the epithelial cells would not be able to produce their own functional versions of XPA.
References
Ensembl Genome Browser [http://www.ensembl.org/index.html]
Goodsell DS. The molecular perspective: Ultraviolet light and pyrimidine dimers. Stem Cells 2001;19(4):348-9.
Human Interactome Map. [http://www.himap.org/]
Kraemer KH, M.D., DiGiovanna JJ, M.D.,Moshell AN, M.D., Tarone RE, Ph.D., and Peck GL, M.D. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin The New England Journal of Medicine 1988;318:1633-1637.
Kohn, Molec. Biol. Cell 1999 Aug;10(8):2703-34
Lin P, M.D., and English JCIII, M.D. Topical treatment of xeroderma pigmentosum Continuing Medical Education 2004;29(8):512-514.
NCBI Conserved Domain Database (CDD) [http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml]
Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 2003 Oct 1;22(19):5293-303.
Thorel F, Constantinou A, Dunand-Sauthier I, Nouspikel T, Lalle P, Raams A, Jaspers NG, Vermeulen W, Shivji MK, Wood RD, Clarkson SG. Definition of a short region of XPG necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol Cell Biol 2004 Dec;24(24):10670-80