SURF1 and Leigh Syndrome: The Role of SURF1 in the Assembly of Cytochrome c Oxidase
Loss of function mutations in the SURF1 gene are associated with the development of Leigh Syndrome, a severe neurometabolic disorder characterized by deficiency of cytochrome c oxidase. The SURF1 gene is a chaperone protein responsible for aiding in the formation of COX assembly intermediates. SURF1 gene is part of a tightly clustered group of genes on chromosome 9, and the genomic organization of these genes have been conserved throughout more than 600 million years of divergent evolution. The SURF1 conserved domain is highly conserved among eukaryotes, highlighting its importance in the respiratory chain. SURF1 has two predicted transmembrane regions associated with alpha helices. Expression analysis of the yeast ortholog of SURF1, SHY1, do not reveal coexpression with COX1, which it is predicted to chaperone. However, SHY1 was more closely regulated with COX18 and TIM44, also located in the inner mitochondrial membrane. COX18 and TIM44 are encoded in the nucleus, and COX1 is located in the mitochondria, which could explain the lack of coregulation between SHY1 and COX1. SURF1 is predicted to interact with Sco2. Both of these proteins are involved in the chaperoning of specific assembly intermediates in COX assembly. One single nucleotide polymorphism located at amino acid 124 produces an unstable protein product, which is not able to accurately chaperone these intermediates, leading to Leigh Syndrome. In order to better understand and treat Leigh Syndrome, more research needs to be conducted to characterize the precise nature of SURF1 activity in chaperoning these assembly intermediates. Because of the dire nature of Leigh Syndrome, gene therapy offers the best possibility for direct treatment of the disease.
Background Information
Diseases associated with dysfunctional mitochondria can be especially dire. The energy requirements of mammalian species depend fully on ATP produced through oxidative phosphorylation, a process localized within the inner mitochondrial membrane (Pecina et al 2004). Cytochrome c oxidase (COX) is the terminal enzyme in the electron transport chain in the mitochondrial membrane (Pecina et al 2004). This enzyme is responsible for coupling electron transport from cytochrome c to oxygen (producing water) by transporting protons from the matrix to the cytosol. Diseases assocatiated with COX are early-onset (symptoms begin to manifest within the first couple of months of life) and are extremely severe (OMIM, Pecina et al 2004).
Leigh Syndrome is one such disease associated with COX deficiency (OMIM, Pequignot et al 2001). The cause of this disease has been localized to mutations in chaperone proteins residing within the nucleus (OMIM, Pequignot et al 2001). The development of Leigh Syndrome is most often associated with loss of function mutations within the SURF1 gene (OMIM, Pequignot et al 2001). The SURF1 protein is located within the mitochondrial membrane and is involved in the biogenesis of integral subunits within COX (Entrez Gene).
Leigh Syndrome is a severe, early-onset neurometabolic disorder characterized by abnormal functioning of the central nervous system and caused by deficient cytochrome c oxidase (COX) biogenesis in the mitochondria (OMIM). The disease is autosomally recessive-inherited, rare, and fatal (OMIM). The disease promotes the creation of bilateral lesions in brain stem tissue, cerebellum, spinal cord, and other areas of the central nervous system (OMIM). Symptoms of the disease include dysphagia, muscle hypotonia, developmental delay, nystagmus, and optic disc pallor, among others (OMIM). Treatment methods consist of treatment of secondary symptoms such as seizures, movement disorders, and cardiac complications aimed at slowing the progression of the disease (The Leighs Center Newsletter). There is currently no method of treating the disease directly, and there is no cure (The Leighs Center Newsletter).
This objective of this study was to perform an in-depth analysis of the SURF1 gene structure, expression, and protein interactions in order to better understand the cause and development of Leigh Syndrome.
References
1) Entrez Gene
[http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene]
2) Online Mendelian Inheritance in Man (OMIM)
[http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM]
3) Pecina P, Houstkova H, Hansikova H, Zeman J, Houstek J: Genetic defects of cytochrome c oxidase assembly. Physiol Res 2004. 53: S213-S223.
4) Pequignot MO, Dey R, Zeviani M, Tiranti V, Godinot C, Poyau A, Sue C, Di Mauro S, Abitbol M, Marsac C: Mutations in the SURF1 gene associated with Leigh syndrome and cytochrome C oxidase deficiency. Hum Mutat 2001 17:374-381.
5) The Leighs Center Newsletter
[http://biochemgen.ucsd.edu/mmdc/lcc01.htm#treatment]
The SURF1 Gene
This section locates potential regions of functional significance in the SURF1 gene by 1) identifying regions of high conservation among divergent species, 2) examining SURF1 protein secondary structure and hydrophobicity, and 3) positioning single nucleotide polymorphisms within the context of protein secondary structure.
Genomic Location
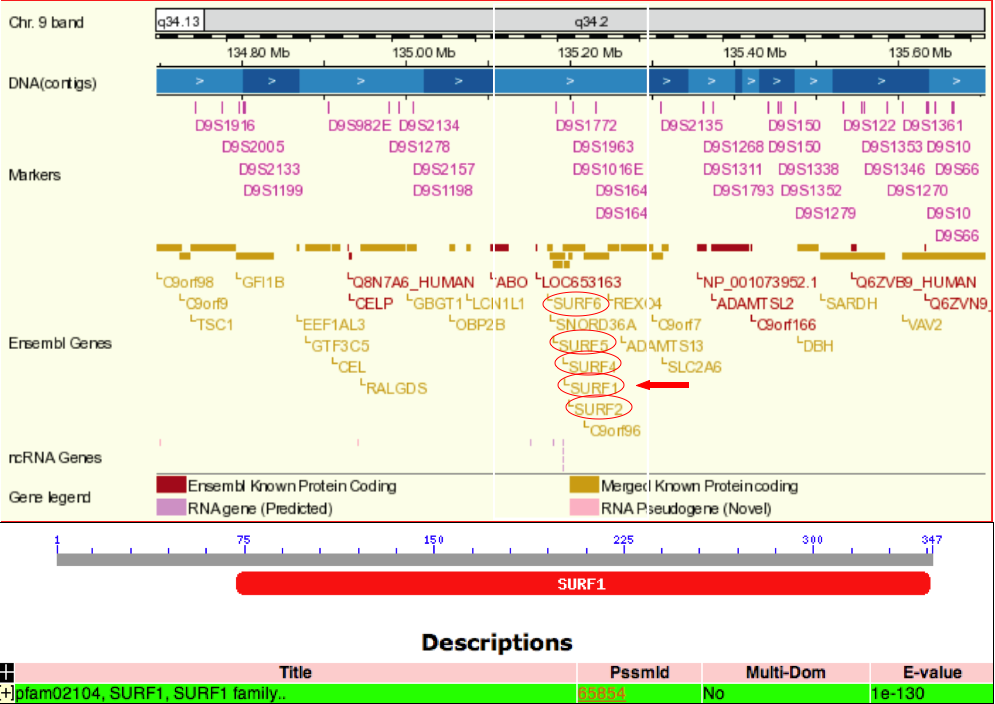
One unique feature of the SURF1 gene is its location in a tight cluster of 6 non-homologous housekeeping genes on chromosome 9q34 (Ensembl Genome Browser). Not only is this cluster one of the tightest in the human genome, it is conserved between humans and birds, representing over 600 million years of divergent evolution (NCBI). The SURF1 gene shares a bidirectional promoter sequence with SURF2 on the opposite strand; these two genes are separated by only 66 base pairs (Ensembl Genome Browser, NCBI). Jaspar analysis of these 66 base pairs revealed one TATA box at a 60% relative profile score threshold on the SURF1 strand (score = -4.360). The function of SURF2 is currently unknown (NCBI). However, the high level of conservation in genomic organization among species suggests that this organization has functional significance.
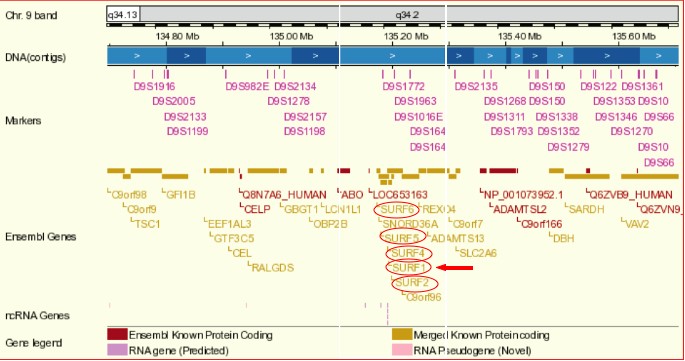
[Figure 1; Click to enlarge]
Figure 1 shows the location of the human SURF1 gene (indicated by the red arrow) on chromosome 9 and its proximity to the SURF housekeeping genes (circled in red). The SURF1 gene has only one conserved domain, which is conserved among eukaryotes, highlighting its importance in the respiratory chain in a wide variety of species.
Conservation Among Species
Orthologs of the SURF1 gene are found in a wide variety of animal species (Ensembl Genome Browser). In order to examine the amount of variation between the SURF1 protein among species, I examined the conservation of SURF1 in humans and 8 divergent animal species (Ensembl Genome Browser). Analysis revealed regions of high conservation among SURF1 orthologs (EBI Tools: ClustalW). Due to the high level of conservation, these regions may be important for the correct functioning of the SURF1 protein.
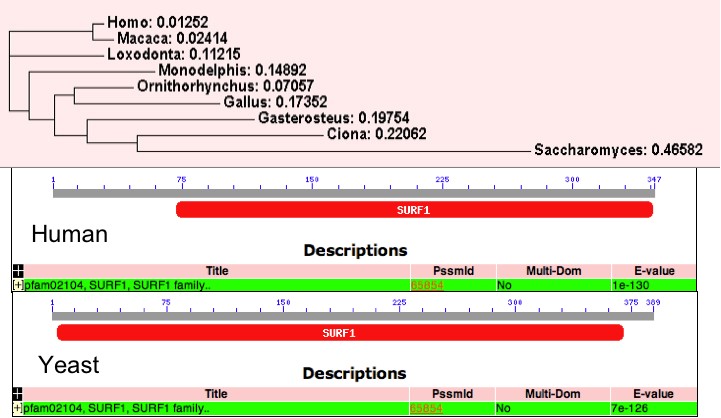
[Figure 2]
Presented in this figure is a phylogram showing the relationship between the SURF1 protein in humans and 8 divergent species and human and yeast conserved domains[Homo = human, Macaca = primate, Loxodonta = African elephant, Monodelphis = possum, Ornithorhynchus = platypus, Gallus = chicken, Gasterosteus = sickleback, Ciona = seasquirt, and Saccharomyces = yeast]. Not surprisingly, the yeast and human orthologs were the most divergent. The human and macaca show the highest level of conservation (96%), while human and yeast show only 27% sequence conservation (EBI Tools: ClustalW).
One key difference between the human and yeast orthologs is a difference in size. SURF1 in humans is 300 amino acids long, while SHY1 (the yeast ortholog) is 389 amino acids long. However, the yeast and human ortholog also share the surfeit 1 conserved domain (human: e-value = 1e-130, yeast: e-value = 7e-126; Figure 2) (CDD). An examination of the gene ontologies of the human and yeast genes show remarkable similarity.
Gene Ontologies (human: Entrez Gene, yeast: SGD)
The gene onotologies of yeast and human are remarkably similar. Both are involved in aerobic respiration and both are located within the mitochondrial membrane. Since these two species are the most divergent evolutionarily, it is likely that SURF1 retains similar function from yeast all the way to humans.
Molecular Function
Human: cytochrome c oxidase acitivity
Yeast: unfolded protein binding
Biological Process
Human: electron transport, COX assembly, aerobic respiration
Yeast: aerobic respiration
Cellular Component
Human: mitochondrion, membrane, integral to membrane
Yeast: mitochondrion, inner membrane, integral to inner membrane
Protein Secondary Structure
Because SURF1 is predicted to be integral to the mitochondrial membrane, transmembrane regions are likely important for the adequate functioning of the SURF1 protein. I used Kyte-Doolittle analyses and TMHMM (2.0) to predict potential transmembrane helices in SURF1. In addition, I used PREDATOR to examine the secondary structure of the SURF1 protein. I predicted that transmembrane regions would coincide with regions of high conservation among species.

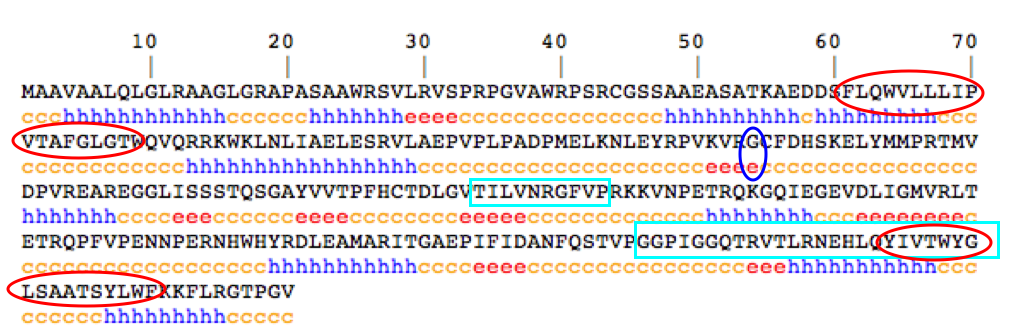
[Figure 3]
This figure shows the secondary structure of the SURF1 protein (EBI Tools: ClustalW, Ensembl, Predator, Uniprot). Black letters represent amino acids, while the colored letters indicate the secondary structure of the amino acid to which it corresponds. The yellow "c" represents a random coil, the blue "h" represents an alpha helix, and the red "e" represents an extended strand. Red circles indicate sections of the protein that the TMHMM (2.0) predicted to be transmembrane regions. Light blue squares represent regions of the highest conservation among species analyzed. The blue circle represents the location of a missense mutation that leads to an unstable SURF1 protein product and the development of Leigh Syndrome.
Kyte-Doolittle analyses revealed no potential transmembrane regions (with a window size of 19 and a threshold of 1.7 indicating hydrophobicity) (Hydropathicity Plots). However, unlike TMHMM, Kyte-Doolittle does not consider secondary structure when predicting potential transmembrane regions. Note in Figure 3 that both transmembrane regions predicted by TMHMM overlap at least partially with alpha helices (score of 25, 18 as a threshold for a transmembrane region). Therefore, protein secondary structure must be considered when locating transmembrane regions within SURF1. In addition, TMHMM analysis of hydrophobicity in the yeast SHY1 ortholog shows two regions of potential transmembrane activity. An analysis of the remaining SURF1 orthologs reveal that each species has at least one potential transmembrane region (TMHMM).
The single nucleotide polymorphism represented by the blue circle in the above figure represented amino acid number 124 (Ensembl, Pequignot et al 2001, Poyau et al 2000, Uniprot). In normal genes, this amino acid is Glycine, a small amino acid which has no charge and is not hydrophilic (AA Properties). The mutation which causes this amino acid to be changed to Glutamate leads to an unstable SURF1 protein product. Glutamate is a large, polar amino acid (AA Properties). Although amino acid 124 is not in a region of highest conservation among species, this particular amino acid had 100% sequence conservation among all species analyzed (ClustalW). Therefore, the small extended strand in which this amino acid is located may be very significant for the correct functioning of the SURF1 protein. This is one of the most common missence mutation leading to Leigh Syndrome (OMIM). Other common mutations that lead to Leigh Syndrome are insertions and deletions that lead to early stop codons and truncated protein products (OMIM).
Conclusions
- Highly divergent species share regions of high conservation that may provide insight into structurally important sections of the SURF1 protein.
- The similarity in gene ontologies and conserved domains of yeast and human suggest that SURF1 function has been largely conserved between yeast and humans.
- Because all species exhibited at least one transmembrane section in the SURF1 protein, location in the membrane is may be necessary for correct functioning of SURF1.
References
1) Amino Acid Properties
[http://www.mcb.ucdavis.edu/courses/bis102/AAProp.html]
2) EBI Tools: ClustalW
[http://www.ebi.ac.uk/Tools/clustalw/index.html]
3) National Center for Biotechnology Information (NCBI)
[http://www.ncbi.nlm.nih.gov]
5) NCBI Conserved Domain Database (CDD)
[http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml]
4) Ensembl Genome Browser
[http://www.ensembl.org/index.html]
5) Entrez Gene
[http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene]
6) Hydrophathicity Plots
[http://www.vivo.colostate.edu/molkit/hydropathy/index.html]
7) Online Mendelian Inheritance in Man (OMIM)
[http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM]
8) Pequignot MO, Dey R, Zeviani M, Tiranti V, Godinot C, Poyau A, Sue C, Di Mauro S, Abitbol M, Marsac C: Mutations in the SURF1 gene associated with Leigh syndrome and cytochrome C oxidase deficiency. Hum Mutat 2001 17:374-381.
9) Poyau A, Buchet K, Bouzidi MF, Zabot M-T, Echenne B, Yao J, Shoubridge EA, Godinot C: Missense mutations in SURF1 associated with deficient cytochrome c oxidase assembly in Leigh syndrome patients. Hum Genet 2000, 106:194-205.
10) PREDATOR
[http://bioweb.pasteur.fr/seqanal/interfaces/predator-simple.html]
11) Saccharomyces Genome Database (SGD)
[http://www.yeastgenome.org/]
12) TMHMM Server v. 2.0
[http://www.cbs.dtu.dk/services/TMHMM/]
13) Uniprot (The Universal Protein Resource)
[http://www.pir.uniprot.org/]
Expression Analysis of SHY1 (yeast ortholog of SURF1)
This section examines the gene expression of the yeast ortholog of SURF1, SHY1. As mentioned in the previous section, the yeast SHY1 shares regions of high conservation with the human SURF1 (EBI Tools: ClustalW). SHY1 also has similar gene ontology with SURF1 (Entrez Gene, SGD). Both genes have the same conserved domain (refer to Figure 2 of previous section) (CDD). Yeast provides an excellent model organism with which to study the expression of SURF1. Yeast has a very short generation time and can be easily placed under different envirnomental conditions in a laboratory setting.
Hoope’s Lab Data
We obtained data from yeast microarrays conducted in the Hoopes Lab at Washington University in St. Louis. The yeast microarrays examined the expression of over 6,000 yeast genes in different growth environments and with deletions of specific genes. I chose to examine the expression of SHY1: 1) in log versus stationary phase, 2) with snf1 deletions in the log versus stationary phase, and 3) with snf1 deletion in the stationary phase versus wild type stationary phase. Below is a summary of these three experiments.
3 Experiments:
1) Log growth vs. stationary growth
- Cy3 = wild type yeast, log phase growth
- Cy5 = wild type yeast, stationary phase growth
2) Snf1 deletion mutant: stationary vs. log
- Cy3 = snf1 deletion mutant, log phase
- Cy5 = snf1 deletion mutant, stationary phase
3) Snf1 deletion mutant vs. wild type
- Cy3 = wild type, stationary phase
- Cy5 = snf1 deletion, stationary phase
I chose to examine the expression of SHY1 in log versus stationary phase for 2 primary reasons. Firstly, yeast has different metabolic requirements in these different stages of growth. Secondly, this experiment provided a control with which to compare my second experiment, the expression of SHY1 with the snf1 deletion. Because SHY1 is involved in the production of COX, which is involved in the production of ATP, determining the difference in expression of SHY1 in different growth environments could provide useful information on the expression pattern of SHY1 under different metabolic conditions. Snf1 regulates transcriptional changes related to glucose depletion (SGD). Because SHY1 is required for aerobic respiration, I predicted that snf1 may cause the expression of genes that prevent the expression of SHY1. I predicted SHY1 to be more highly expressed in the absence of snf1. Because SHY1 is thought to be involved in the biogenesis of COX1, I wanted to track the expression of SHY1 with COX1 under diferent experimental conditions (SGD). COX1 gene is located in the mitochondria (SGD).
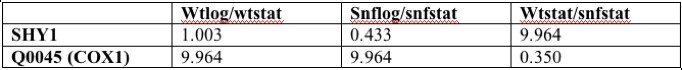
[Figure 1]
Figure 1 shows the expression values (log-transformed) of SHY1 and COX1 in the 3 experiments. Notice that SHY1 and COX1 do not show co-regulation within these experiments. SHY1 does not drastically change expression between log and stationary phase with or without the snf1 deletion. However, SHY1 was heavily induced in cells with the snf deletion versus wild type. This follows the prediction that the presence of snf1 dampens the expression of SHY1 either through direct repression, or, more likely, through the expression of genes that repress SHY1. COX1 is highly induced when in stationary phase versus log phase growth (with or without the snf1 deletion). The snf1 deletion did not cause significant induction or repression of the gene in the third experiment.
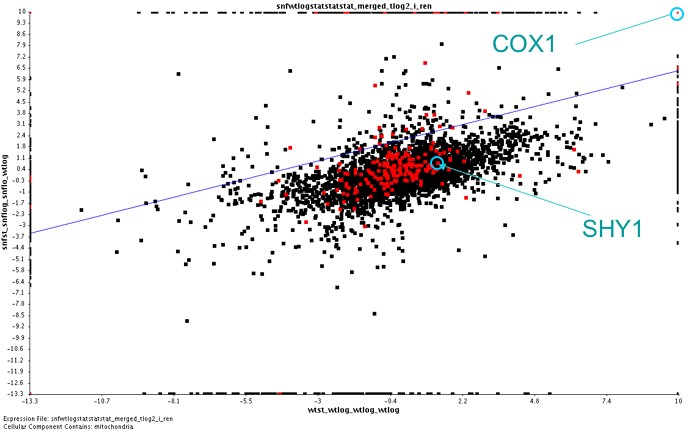
[Figure 2; click to enlarge]
Figure 2 shows a pairwise comparison of all the yeast genes in experiments 1) log versus stationary wildtype (x-axis) and experiment 2) log versus stationary snf1 deletion (y-axis). SHY1 and COX1 expressions are labelled in light blue. Red points indicate genes which are associated with the mitochondrion (cellular component). Notice that mitochondrial genes seem to be evenly spread in this pairwise comparison, indicating that there was not uniform induction or repression of mitochondrial genes within the different experimental conditions.
I used heirarchical clustering (threshold 0.0001) to determine which genes were expressed similarly to SHY1. I found a total of 64 genes that clustered with SHY1. Two of the 64 genes were located within the mitochondria: TIM44 and COX18. TIM44 is located on chromosome 9 and is involved in protein transport (SGD). The human ortholog has the same name and function (Ensemble, Entrez). COX18 is located on chromosome 7 and is required for the export of COX2 protein from the mitochondrial matrix into COX enzyme complex (SGD). In humans, COX18 is also involved in the transport of proteins into the mitochondrial membrane, but not specifically COX2 (Ensembl, Entrez).
I looked to the Saccharomyces Genome Database to answer the following questions:
Is the expression of SHY1 and COX1 correlated in different conditions related to metabolism?
How does the expression of TIM44 or COX18 in different environmental conditions follow the expression of SHY1?
How does snf1 expression relate to SHY1 expression?
Saccharomyces Genome Database - Expression Connection

[Figure 3]
The top experiment in Figure 3 shows the expression of SHY1, COX1, and the 2 genes of interest as determined through Hoopes Lab microarray experiments (Lai et al 2006, SGD). This microarray experiment explores the expression of these genes in response to anoxia and subsequent reoxygenation in glucose (Lai et al 2006). Reoxygenation began at the 6.00 time point (Lai et al 2006). Because oxidative phosphorylation requires oxygen, the repression of these genes in an anoxic environment is not surprising. Interestingly, SHY1, TIM44, and COX18 are repressed to a lesser degree than COX1. This could be explained by the fact that the SHY1, TIM44, and COX18 are all located in the nucleus, while COX1 is located in the mitochondria. These genes may therefore be regulated differently. SHY1 has a Pearson correlation of greater than or equal to 0.8 with all three of the other genes (Figure 3).
The second experiment in Figure 3 shows the expression of SHY1 and snf1 during the unfolded protein response (SGD, Travers et al 2000). Because both genes are induced during this experiment, the data suggest that the snf1 does not indirectly repress SHY1. Because both the SHY1 protein and the snf1 protein act as chaperones, induction during the unfolded protein response is not surprising.
Future Research
The TRIPLES database provides information on the reaction of yeast cells when certain genes are mutated in different experimental conditions. When SHY1 was mutated, yeast cells were strongly affected in the condition in which only glycerol was present (TRIPLES). In bacteria, glycerol prevents fermentation, forcing the yeast cells to undergo cellular respiration for energy (Cellular Respiration). This condition was tested in order to locate genes involved in cellular respiraton.
One future microarray experiment would be to track the expression of SHY1, COX1, COX18, and TIM44 under different levels of glucose availability. If glucose presence influences the expression of these proteins at all, I would predict that these proteins would be induced in high glucose environments and repressed in low glucose environments. This experiment could also test differences in expression of mitochondrially-encoded COX subunits (such as COX1) and nuclearly-encoded COX subunits (such as COX18).
Design for Proposed Experiment
1) Grow cells with differing amounts of glucose availability
2) Experimental microarray analysis:
- Cy3 = glucose average availability
- Cy5 = glucose depletion OR glucose excess
3) Platform: all nuclear and mitochondrial yeast genes
4) Controls:
- Same availability vs. same availability
- - Controls for stochasticity
- Dye swap control
- - Controls for transcript-dependent dye bias
Conclusions
- SHY1 expression is not highly correlated with COX1 expression in the experiments considered. Possible explanations for this include 1) that SHY1 and COX1 are regulated differently due to differences in genomic location (nucleus versus mitochondria) or 2) that SHY1 is involved in chaperoning other proteins besides COX1 and therefore is not solely regulated with COX1.
- Snf1 may play a role in SHY1 repression; however, other regulatory processes may be involved this regulation.
References
1) Cellular Respiration
[http://www.biology.iupui.edu/biocourses/N100/2k4ch7respirationnotes.html]
2) EBI Tools: ClustalW
[http://www.ebi.ac.uk/Tools/clustalw/index.html]
3) Ensembl Genome Browser
[http://www.ensembl.org/index.html]
4) Entrez Gene
[http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene]
5) Lai LC, Kosorukoff AL, Burke PV, Kwast KE: Metabolic-state-dependent remodeling of the transcriptome in response to anoxia and subsequent reoxygenation in Saccharomyces cerevisiae. Eukaryot Cell 2006. 5:1468-8149.
6) NCBI Conserved Domain Database (CDD)
[http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml]
7) Saccharomyces Genome Database (SGD)
[http://www.yeastgenome.org/]
8) Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P: Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000,101:249-58
9) TRIPLES
[http://ygac.med.yale.edu/triples/default.htm]
Leigh Syndrome Disease Model
Potential Protein Interactions
While SURF1 is known to be a chaperone protein aiding in the assembly of COX, the exact function of the SURF1 gene is unknown (Stiburek et al 2005). The Human Interactome Map predicts three proteins to interact with SURF1 (Human Interactome Map). Sco2, also located in the mitochondrial membrane, is involved in chaperoning the same COX assembly intermediate as SURF1 (Entrez Gene, Stiburek et al 2005). Mutations in Sco2 also lead to the development of Leigh Syndrome (Pecina et al 2004, Stiburek 2005). However, Sco2 mutations are more rare than SURF1 mutations (Pecina et al 2004). APCS is not localized to the mitochondrial membrane and is thought to play a role in the degradation of chromatin (Entrez Gene). It is also thought to be involved in binding to apoptotic cells (Entrez Gene). Because cytochrome c is released from the mitochondrial membrane during apoptosis, the interaction between SURF1 and APCS probably only occurs during the process of apoptosis (Reproductive and Cardiovascular Disease Research Group). SDHA is localized within the mitochondrial membrane and is involved in the assembly of succinate-ubiquinone oxidoreductase, another complex in the respiratory chain (Entrez Gene). Mutations in this gene may also lead to Leigh Syndrome (Pecina et al 2004). Of these three genes, Sco2 is the most likely to be directly involved in chaperoning proteins with SURF1 under normal conditions.
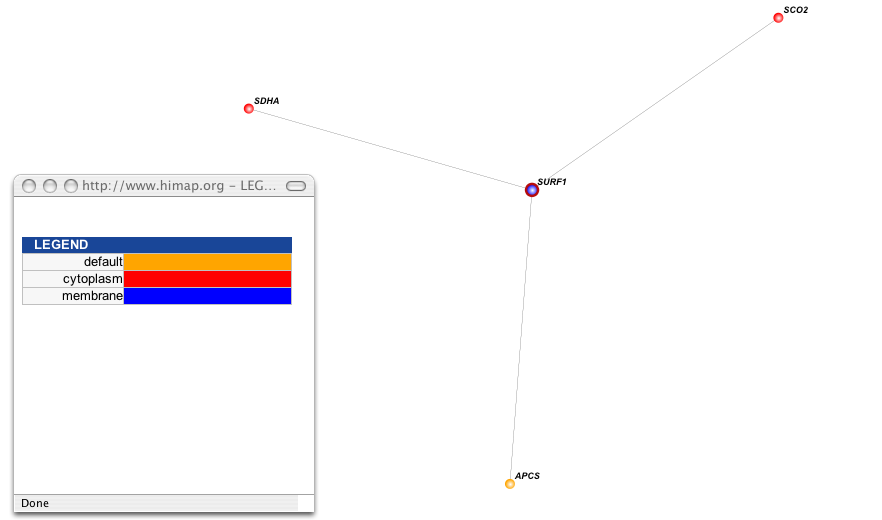
[Figure 1; click to enlarge]
Circuit Diagram: Wild-type Versus Diseased
In mammals, COX consists of 13 subunits, which can be either mitochondrially or nuclearly encoded (Pecina et al 2004). The 3 mitochondrially-encoded proteins are the largest, most ancestral of the subunits and form the catalytic core of the enzyme (Pecina et al 2004). The nuclearly-encoded proteins are smaller and are involved in the regulation and assembly of the enzyme as a whole (Pecina et al 2004). The biogenesis of COX occurs systematically with the sequential incorportation of subunits from both the cytosol (nuclearly-encoded) and the mitochondrial matrix (mitochondrially-encoded). Because some of these steps are rate-limited, 4 assembly intermediates are formed (Pecina et al 2004).
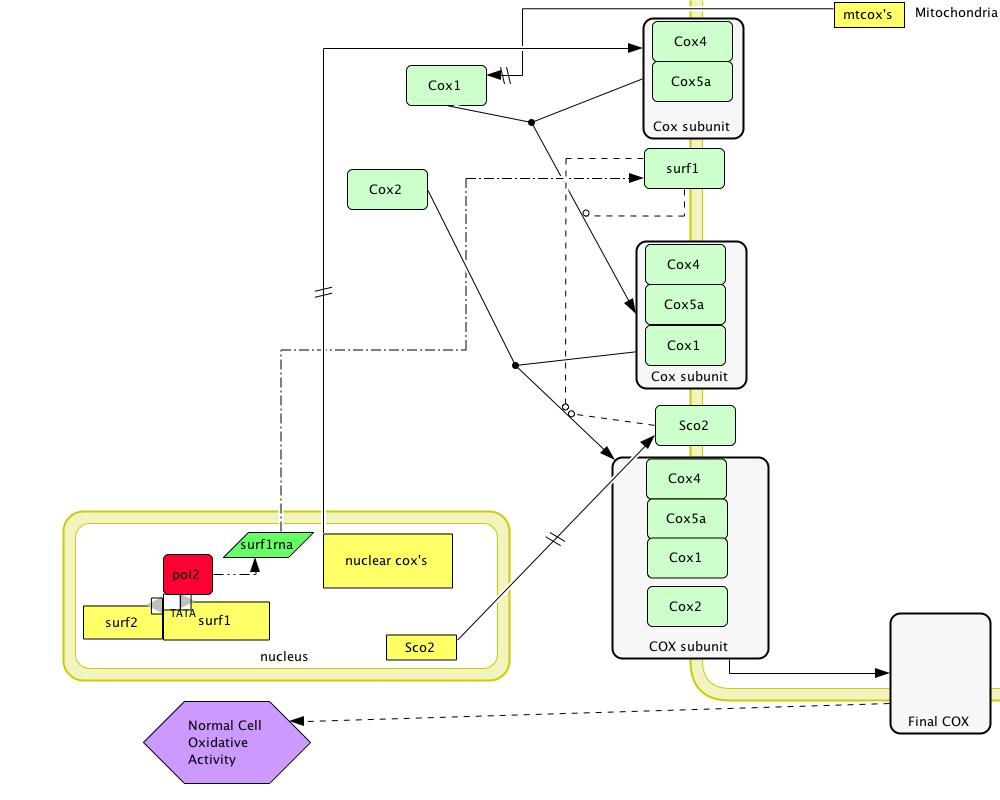
SURF1 acts as a chaperone protein involved in biogenesis of the 2nd and 3rd assembly intermediates in the formation of COX (Pecina et al 2004, Stiburek et al 2005). These two intermediates are composed of proteins encoded in both the nucleus and the mitochondria (Pecina et al 2004, Stiburek et al 2005). The exact role of the SURF1 protein in chaperoning these subunits within humans is currently unknown (Pecina et al 2004, Stiburek et al 2005). However, it has been proposed that SURF1 is involved in the biogenesis of the mitochondrially-encoded COX1 protein, and is potentially involved in chaperoning assembly intermediates in which COX1 is a constituent (Stiburek et al 2005). COX1 forms an assembly intermediate with COX4 and COX5a, both encoded in the nucleus (Stiburek et al 2005). Studies have shown that COX4 and COX5a form a heterodimer before being joined with COX1 (Stiburek et al 2005). Therefore, SURF1 is predicted to chaperone the creation of the assembly intermediate consisting of COX4a, COX5, and COX1. This is the 2nd assembly intermediate in the formation of the complete COX enzyme (Pecina et al 2004, Stiburek et al 2005).
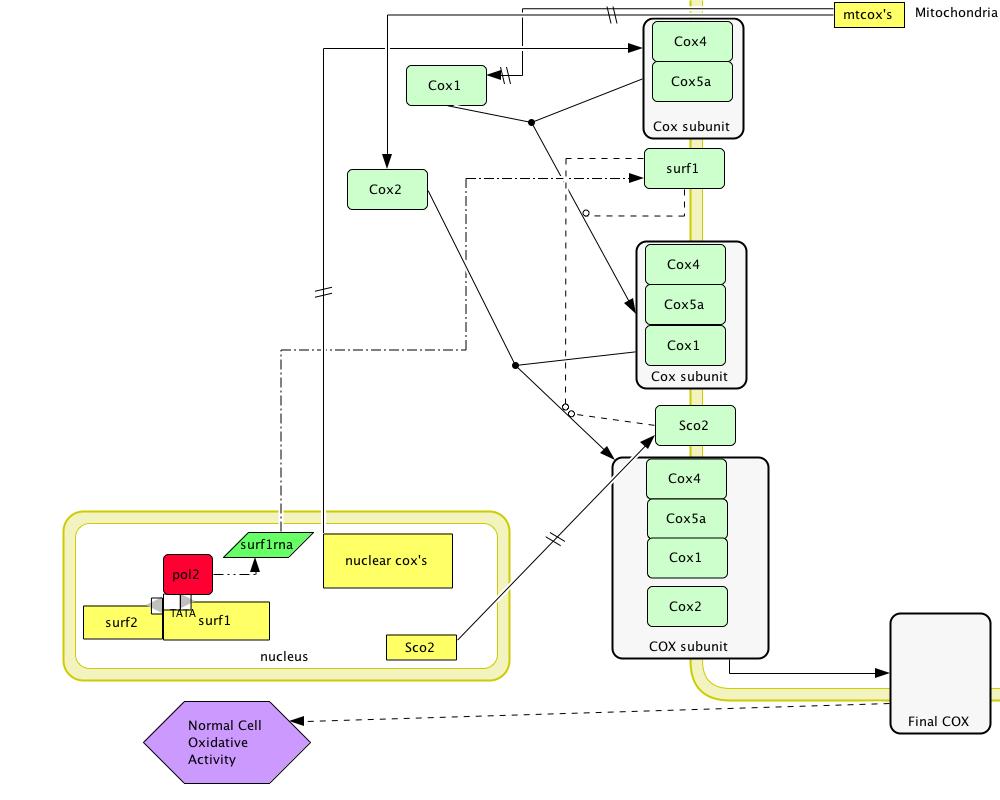
[Figure 2; click to enlarge]
Figure 2 shows the normal model for SURF1 function. Green squares with rounded edges are proteins. Yellow squares indicate genes. SCO2 chaperones the creation of the 3rd assembly intermediate containing COX4, COX5a, COX1, and the mitochondrially-encoded COX2 by aiding in the addition of the Cu to the center of the COX2 subunit (Stiburek et al 2005). This Cu center stabilizes the COX2 subunit (Stiburek et al 2005). Because the Human Interactome Map predicted that SURF1 interacts with SCO2, it is likely that SURF1 is also involved in the assembly of this intermediate (Human Interactome Map).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
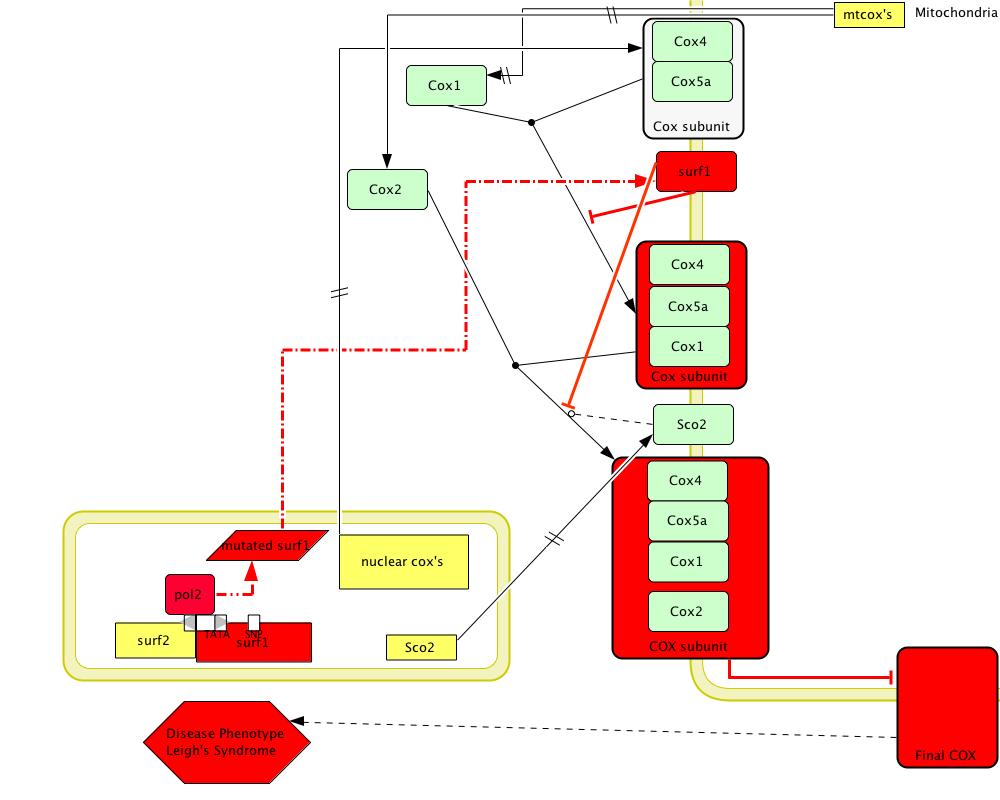
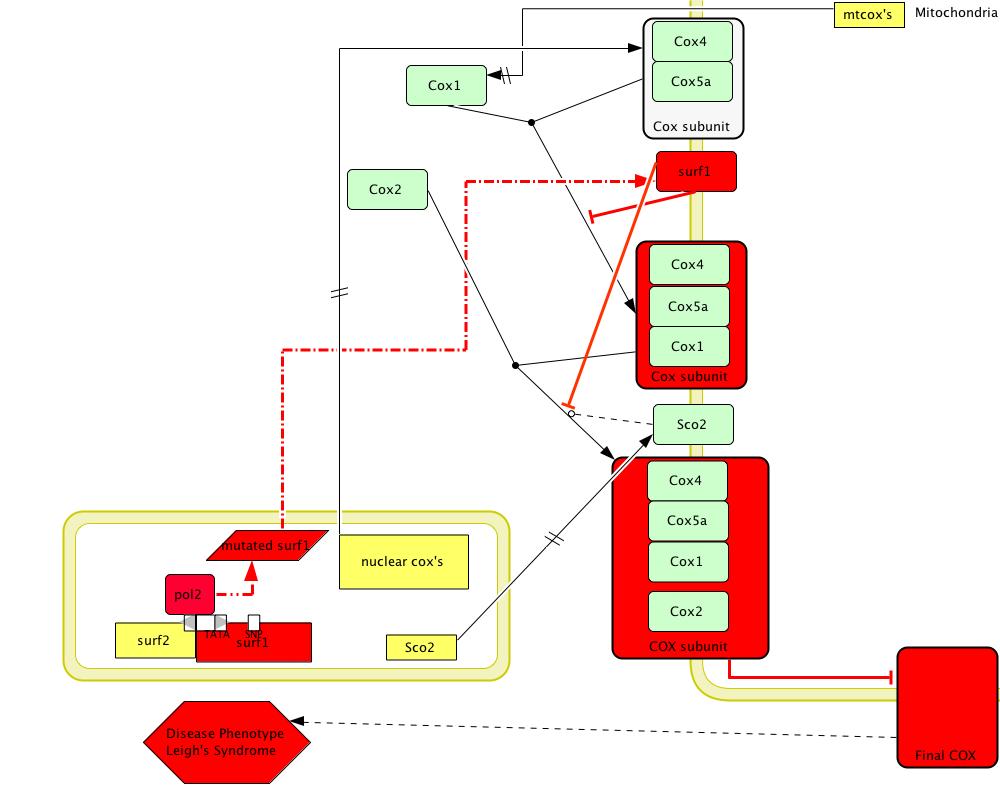
[Figure 3; click to enlarge]
Figure 3 shows a model of the disease phenotype of SURF1 due to a missense mutation which changes amino acid 124 from Glycine to Glutamate (Pequignot et al 2001, Poyau et al 2000). Red colored shapes show mutated or non-functional products. This missence mutation creates an unstable protein product which is not able to act as a chaperone and therefore produces an unstable COX enzyme (Pequignot et al 2001, Poyau et al 2000). The inability to undergo oxidative phosphorylation leads to Leigh Syndrome.
Future Experiments
Although it is fairly well known that SURF1 is involved in chaperoning COX subunits, the exact action of SURF1 in the assembly of these intermediates remains unknown (Pecina et al 2004, Stiburek et al 2005). The circuit diagrams presented above could be both verified and complemented by further research on the exact nature of SURF1 chaperoning activity. A series of immunoprecipitation experiments may shed light on the precise interactions of the SURF1 gene. One such experiment would include precipitating COX1 from a healthy cell and testing for the presence of predicted bound proteins in a complex with COX1 using a western blot. I would expect to find COX1 bound to COX2, COX4, and COX5a.
By comparing the results of this experiment with the same experiment with mutated SURF1, I could localize the precise step that the mutation caused in chaperone deficiency. For instance, if I found that COX1 was bound to COX4 and COX5a, I would conclude that SURF1 was able to chaperone the first assembly intermediate but not the second (addition of COX2). If I found no proteins bound to COX1, I would conclude that SURF1 was not able to chaperone any assembly intermediate.
By creating mutations in functional regions of SURF1, such as a transmembrane region or those regions highly conserved among animal species, and running an immunoprecipiation experiment, I could definitively locate regions of the protein that are functionally significant. Additionally, I could perform an immunoprecipitation experiment with a healthy cell, but precipate SURF1. I could then use mass spectrometry to identify proteins not previously predicted to bind to SURF1. These experiments would aid in the understanding of SURF1 chaperoning activity and may help in treating those afflicted with Leigh Syndrome.
Potential Treatments
Leigh Syndrome is a fatal, fast-acting disease which can cause severe mental and physical damage in a short amount of time (OMIM). There are currently 30 documented mutations in the SURF1 gene which can lead to the development of Leigh Syndrome (OMIM). All of these mutations code for a truncated protein or an unstable protein product (as with the mutation depicted in the above circuit diagram) (OMIM). Prevention of the disease through pre-natal screening or in vitro fertilization is a possibility for individuals who are at risk of being heterozygous for the disease allele. However, due to the rarity of the disease, it is likely that an individual heterozygous for a disease allele would not be aware this possibility and would be unlikely to be testsed. Therefore, direct treatment methods need to be developed in order to increase the likelihood of survival of children born with this disease.
Because death can occur as quickly as just a few months after the beginning of symptoms, treatment of the disease must be fast and effective. Treatment of secondary symptoms should begin as soon as symptoms begin in order to prolong life until a direct form of therapy can be instituted. Due to the overall severity of COX defects caused by Leigh Syndrome, it is unlikely that a drug could be developed for effective treatment of this disease. Gene therapy seems to be the most promising potential treatment of Leigh Syndrome.
The use of protein carriers as a delivery method for gene therapy may be effective. Because Leigh Syndrome is a recessive disease and non-functional SURF1 protein is not predicted to actively disrupt any other cellular process, the addition of a wild-type SURF1 gene in vivo could prove effective in restoring functional COX to the inner mitochondrial membrane. Although there are certain risks involved in transporting proteins into tissues as sensitive as brain tissue, the severity of the disease justifies some risks in treatment.
Conclusions
- The SURF1 chaperone protein is integral to the correct formation of COX.
- Future experiments that investigate the nature of SURF1 function will aid in understanding Leigh Syndrome
- Because of the importance of the COX enzyme and the complicated nature of COX assembly, gene therapy of SURF1 seems to be the best treatment method.
References
1) Entrez Gene
[http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene]
2) Human Interactome Map
[http://www.himap.org/]
3) Online Mendelian Inheritance in Man (OMIM)
[http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM]
4) Pecina P, Houstkova H, Hansikova H, Zeman J, Houstek J: Genetic defects of cytochrome c oxidase assembly. Physiol Res 2004. 53: S213-S223.
5) Pequignot MO, Dey R, Zeviani M, Tiranti V, Godinot C, Poyau A, Sue C, Di Mauro S, Abitbol M, Marsac C: Mutations in the SURF1 gene associated with Leigh syndrome and cytochrome C oxidase deficiency. Hum Mutat 2001 17:374-381.
6) Poyau A, Buchet K, Bouzidi MF, Zabot M-T, Echenne B, Yao J, Shoubridge EA, Godinot C: Missense mutations in SURF1 associated with deficient cytochrome c oxidase assembly in Leigh syndrome patients. Hum Genet 2000, 106:194-205.
6) Reproductive and Cardiovascular Disease Research Group
[http://www.sgul.ac.uk/depts/immunology/~dash/apoptosis/mito.htm]
7) Stiburek L, Vesela K, Hansikova H, Pecina P, Tesarova M, Cerna L, Houstek J, Zeman J: Tissue-specific chytochrome c oxidase assembly defects due to mutations in SCO2 and SURF1. Biochem J 2005. 392: 625-632.