*This webpage was produced as part of an undergraduate course at Davidson College*
Paper Review
Coat Variation in the Domestic Dog Is Governed by Variants in Three Genes
Edouard Cadieu, Mark W. Neff, Pascale Quignon, Kari Walsh, Kevin Chase, Heidi G. Parker, Bridgett M. VonHoldt, Alison Rhue, Adam Boyko, Alexandra Byers, Aaron Wong, dana S. Mosher, Abdel G. Elkahloun, Tyrone C. Spady, Catherine Andre, K. Gordon Lark, Michelle Cargill, Carlos D. Bustamante, Robert K. Wayne, Elaine A. Ostrander
Science, Vol. 326: 150-3. 2 October 2009. (Link)
Summary:
In this paper, the researchers present data from multiple genome wide association studies (GWAS) investigating genes associated with canine fur phenotypes. Specifically, they focused on three characteristics of the canine coat (pelage): (i) presence or absence of furnishings [growth pattern of moustache and eyebrows typically observed in wire-haired dogs], (ii) hair length, and (iii) the presence or absence of curl . To investigate this, they produced three genome-wide single nucleotide polymorphism (SNP) data sets comprised of: 96 dachshunds segregating three coat varieties (wire-haired with furnishings, smooth, and long-haired without furnishings), 76 Portugese water dogs segregating the curl phenotype, and 903 dogs from 80 breeds (termed CanMap) exhibiting a large spectrum of phenotypes. Even though the genetic basis of coat color has been well estabolished, little was known bout the genes involved in the numerous phenotypic variants observed in dog breeds. This study identified distinct mutations in three genes involved in coat phenotypes (RSPO2, FGF5 & KRT71) that accounted for most purebreed dogs in the United States. Considering that "most [dog] breeds likely originated within the past 200 years, [the] results [from this study] demonstrate how a remarkable diversity of phenotypes can quickly be generated from simple genetic underpinnings." The investigation presented by Cadieu et al., offers a novel strategy for enhancing the genetic understanding of a complex phenotype and provides a new way in which the evolution of mammalian form and function could be viewed.
Method:
All three traits of interest were investigated using the same method:
1) a GWAS for a specific trait (hair length, presence/absence of curl, etc.) was preformed within a specific breed separating the phenotype of interest to identify the strongest associated locus with this phenotype.
2) a second GWAS using the CanMap data set was preformed to again identify highly associated loci. This second assesment allows for the user to rule out false-positives associated with the phenotype due to population structure within the breed.
3) Fine-mapping of homozygous regions was used to determine the smallest shared haplotype between the two previous GWASs.
4) Finally sequencing of the locus was preformed to identify mutations resulting in varied phenotypes.
Figure 1: RSPO2 identified as the associated gene for moustache and eyebrow growth patterns (furnishings).
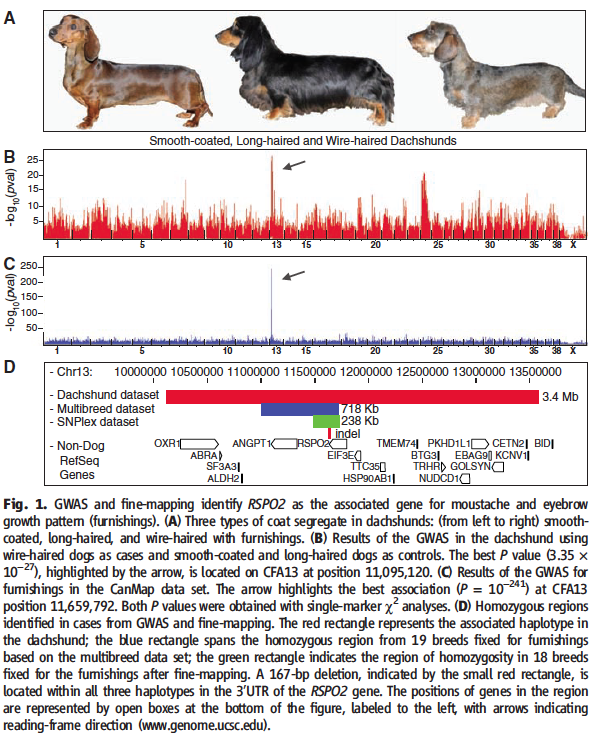
A) This box illustrates three coat types segregated in dachshunds. The smooth-coated (left) and the long-haired (middle) were used as controls and wire-haired with furnishings (right) as cases for the GWAS and CanMap analyses. This part of the figure was illustrated as a reference for the described coat phenotypes analyzed in this study.
B) GWAS results from the dachshund data set. The genome is plotted on the x-axis, with numbers indicating the chormosome, and the negative log of the p value is plotted on the y-axis. This transformation allows the researchers to identify which association least likely occured due to chance and thereby can draw connections between variance observed and the phenotypic trait. Single-marker analysis of the dachshund GWAS data and linkage analysis of the pedigree identified the same locus on chromosome 13 (CFA13), as indicated by the arrow (best p value).
C) GWAS results from the CanMap data set. Again, the arrow highlights the best p value which is located on CFA13, confirming results from 1B. For both 1B and 1C, peaks were determined by single-marker χ² analysis. Interestingly, the peak value at CFA13 was much greater for Figure 1C than in 1B. This could be due to the CanMap data set having sampled nearly 10x as many dogs as the dachshund data set. It is also interesting to note that the high association peak just before CFA25 seen in the dachshund GWAS does not appear in the CanMap--indicating CFA13 as the locus best associated with the trait.
D) This panel summarizes the steps imployed to isolate the homozygous region identified in cases from GWAS and fine-mapping. As described in the figure legened, the different colored rectangles indicate each reduction in homozygous region (determined by methods 1-4). Fine-mapping reduced the homozygous region to 238kb. This was determined to span the RSPO2 gene (excluding the 5' UTR and the first exon). The small red rectangle indicates a 167-bp insertion in the 3' UTR of the RSPO2 gene. This 167-bp fragment was perfectly associated with the furnishings trait in dogs (case/control study & pedigree analysis). This was confirmed in a comparison of 704 dogs with varying phenotypes. Out of 298 dogs with furnishings, 268 were homozygous and 29 were heterzygous for this insertion. 406 dogs lacking the trait were determined as being homozygous for the ancestral state--indicating a dominant mode of inheritance for this insertion.
The authors go on to describe RSPO2 in the text as being an excellent candidate for a hair-growth pehnotype. They cite the Wnt/beta-catenin pathway and its involvement in hair follicle establishment. Additionally, they reference this pathway for its involment in hair folicle tumors which occur most frequently in breeds with furnishings.
According to the authors, this mutation does not affect the protein coding region of the RSPO2 gene. However, they speculate that since this region falls in the 3' UTR, that the mRNA stability of the RSPO2 gene may be comprimised. This was confirmed through expression level analysis of the mRNA in muzzle skin biopsies of dogs with furnishings--indicating this mutation as causing transcription problems consistent with their speculations.
Figure 2: Regions of homozygosity identify genes for pelage length and curl.
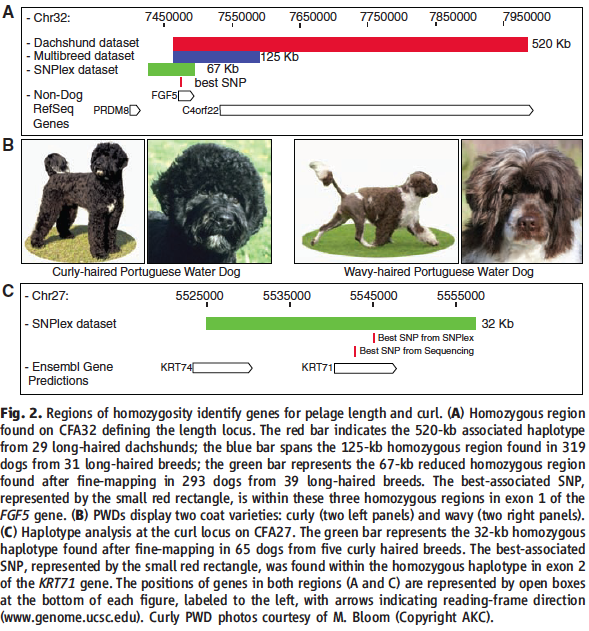
A) This panel is similar to that of the one shown in Figure 1D. It illustrates the summary of steps employed to isolate the length locus on chromosome 32. Fine-mapping revealed a 67-kb homozygous region whose best associated SNP (indicated by small red rectangle) resides within exon 1 of the FGF5 gene. The authors then refer to a previous study that identified the FGF5 gene in Welsh corgis as being associated with long-haired phenotype--this panel replicates this previous finding. Sequencing of their best associated SNP revealed a change in a highly conserved cysteine to phenylalanine in the first exon of the FGF5 gene. Subsequent typing of several hundred dogs with varying hair lengths for this SNP region suggested a recessive mode of inheritance for hair length: all long-haired dogs had the TT genotype while all short or wire-haired dogs had either the GT or GG genotypes.
B) Here the authors again provide pictures of the different phenotypes being investigated. Curly-haired (left) and wavy-haired (right) Portuguese water dogs are shown.
C) This panel presents the haplotype analysis at the curl locus on CFA27. Similar to panel A, here they present results from a GWAS within Portuguese water dogs and a GWAS within the CanMap data set. This time however, they show only the 32-kb fine-mapping region, which uncovered a shared homozygous haplotype that included two keratin genes. Sequencing of this region identified one SNP located in the KRT71 gene resulting in the nonsynonymous modificatoin of arginine to tryptophan. Mutations in KRT71 suggest keratins as being probable candidates for hair growth (previously determined in curly coated mice). The mutation of Arg --> Trp identified here occured in the second exon of the gene and may affect a coiled-coil and/or a prefoldin protein domain. Cadieu et al., suggests that changes in these protein domains could affect cellular targeting, receptor binding, or folding of the protein after translation.
Figure 3: Combinations of alleles at three genes create seven different coat phenotypes.

This figure simply summmarizes the results observed in the previous two figures. The table on the left presents the three major traits both within and across multiple breeds. Here they show that the combinations of these genotype mutations (RSPO2, FGF5 & KRT71) generate at least seven coat types, whereas variuos mutation combinations account for 95% of all dog coat phenotypes observed in the study.
As a reference control, three gray wolves were analyzed for these mutations. No mutations were found in either the gray wolves or the short-haired dogs--demonstrating that short-haired dogs carry the ancestral alleles. The authors then speculate "that a single mutation occured for each trait and was transferred multiple times to different breeds through hybridization."
Conclusion & Critique:
Overall, I thought this paper did an excellent job of examining a complex phenotypic issue. The data and information was presented in a systematic way that was easy to follow and understand. Through the use of GWAS and pedigree linkage analysis, Cadieu et al., clearly demonstrated that they had identified key genes controlling an array of coat phenotypes in domesticated dogs. These combinations of the three identified mutations explained nearly 95% of all observed coat phenotypes in over 1000 dogs from 80 domestic breeds used in the study. This finding surprised me. Even more so was that each trait examined (furnishings, hair length, and curl) corresponded with a single mutation located on only one gene. Despite their convencing data, I am still optimistic to believe that all variants of coat character could be determined by such a small subset of genes. While the authors identified limitations in their study a few aspects of this paper are still bothersome.
First, in the figure legend for Figure 1D, the authors describe this 167-kb homozygous fragment as being a "deletion" within all three haplotypes in the 3' UTR of the RSPO2 gene. However, in the text of the paper, this same fragment is described as an "insertion" on multiple occasions. Although this discrepancy could be a trivial issue caused by an error in typing, it complicates the comprehension of their data. Secondly, only 108 of the 160 recognized dog breeds were used in this study. Therefore, nearly 1/3 of possible breeds were not included in this study. We might expect that with their inclusion, some novel mutations would arise in addition to the possibility of multiple gene interactions occuring--resulting in the large spectrum of coat phenotypes observed in dogs. Finally, the authors acknowledge that some breeds exhibiting the long-haired phenotype (for example) are not caused by the mutations identified in this paper. This makes me believe that other unidentified mutations (or genes for argument sake) could account for similar phenotypes.
All in all, I enjoyed this paper and found it to be quite edifying. It would be interesting to see what directions these authors, or others in the same field, take in order to complete this partial picture of coat color and type for domestic dog breeds.
Reference:
Cadieu, E, Neff, MW, Quignon, P, Walsh, K, Chase, K, Parker, HG, VonHoldt, BM, Rhue, A, Boyko, A, Byers, A, Wong, A, Mosher, DS, Elkahloun, AG, Spady, TC, André, C, Lark, KG, Cargill, M, Bustamante, CD, Wayne, RK, and Ostrander, EA. 2009. "Coat variation in the domestic dog is governed by variants in three genes." Science 326: 150-153.
William's Home Page
Genomics Page
Biology Home Page
Email: William Green for questions or comments.
© Copyright 2011 Department of Biology, Davidson College, Davidson, NC 28035