The ABCD1 Gene &Adrenoleukodystrophy
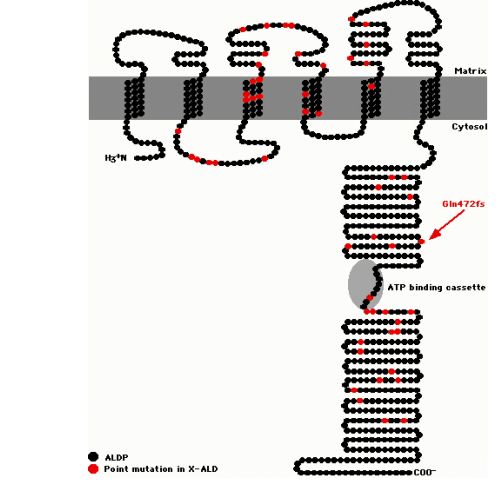
Figure 1:A topological model of the Adrenoleukodystrophy protein (ALDP). Common point mutaions are in red, and the black indicates the protein. Image from.Dr. Celia Kassmann, 2006. Permission Pending.
Overview and History:
The ABCD1 gene (also referred to as the ALD gene) is located on the X-chromosome at position Xq28 (NCBI, 2011). This gene spans 21 kb of genomic DNA, consists of 10 exons, and codes the ALD protein (ALDP) that is made up of 745 amino acids (NCBI, 2011). The ABCD1 gene product is a crucial protein, as it is an ATP–binding cassette (ABC) transporter that aids in the peroxisomal transport of fatty acids (NCBI, 2011). The ALD protein resides in the membrane of the peroxisomes, and it is a half transporter, thus it requires a “partner-half” to become functional (NCBI, 2011). The dimerized, functional ABC transporter then imports fatty acids into the peroxisome, where the fatty acids are catabolized (NCBI, 2011).
A defect in the ABCD1 gene results in a disease called Adrenoleukodystrophy, or ALD (NCBI, 2011). This disease is an X-linked disorder that results in the accumulation of very long chain fatty acids (VLCFA). The accumulation of such fatty acids results in neurological degeneration over time, because the fatty acids demyelinate nerves in the nervous system (Cartier et al., 2009). The degenerative nature of the disease decreases the ability of the nervous system to send and receive nerve impulses, and this deterioration results in strokes, seizures, motor skill loss, and blindness. Death generally occurs 1-10 years after symptoms first appear (NCBI, 2011). The disease is relatively rare as it occurs in about 1 of every 20,000 individuals (NCBI, 2011). Adrenoleukodystrophy has three phenotypes: Adrenomyelopathy, Addison’s type, and Cerebral ALD (Cartier et al., 2009). The former two subsets of ALD manifest themselves later in life (20-35 years of age), and they are the less severe forms of ALD. The cerebral form of ALD is the most common form of ALD, and it appears in early childhood (4-10 years of age). The disease generally occurs in males, because of the disease’s X-linked nature, however females can also be affected. Their symptoms are much more mild than Adrenoleukodystrophy in males, because they are somewhat protected due to the presence of their other X chromosome.
Currently, there are no cures for ALD, but there are a variety of treatments such as bone marrow transplants and Lorenzo’s oil (NCBI, 2011). Lorenzo’s oil is a combination of naturally occurring oils in high doses that delay the rate of fatty acid chain development when taken regularly. The bone marrow transplants are the most effective form of treatment, however they are extremely painful and can cause death. The transplanted bone marrow must be donated from a matched person that has a functional ALD protein. The engrafted bone marrow slows the rate of the neurological deterioration and it has a rehabilitating affect. These positive effects are caused by the engrafted bone marrow's creation of new cells that create functional ALD proteins, which break down the accumulated VLCFAs (NCBI, 2011). Currently, much work is being performed on the treatment of ALD with gene therapy.
References:
Nathalie Cartier, Alima Hacein-Bey-Abina, Cynthia C. Bartholomae, Gabor Veres, Manfred Schmidt, Ina Kutschera, Michel Vidaud, Ulrich Abel, Liliane Dal-Cortivo, Laure Caccavelli, et al. Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy Signal processing in single cells. Science [Internet]. 2009 November 6[cited 2011 Jan 17]; 326(5954): 818-822 . Available from: http://www.sciencemag.org/content/326/5954/818.short
National Center for Biotechnology Information. ABCD1 ATP-binding cassette, sub-family D (ALD), member 1 Homo sapien [internet].Report Series. Bethesda (MD): U.S. National Library of Medicine. 2011 [cited 2011, Jan 19]; [about 17 screens]. Available from: http://www.ncbi.nlm.nih.gov/gene/215
If you have any questions or comments please contact Hunter Hayes