Review: "Coat Variation in the Domestic Dog Is Governed by Variants in Three Genes"
Overview:
In this paper, the researchers wanted to determine the genetic basis that was responsible for the variations in dog coats (pelage). To accomplish this task, the researchers localized the genes that resulted in three pelage traits: furnishings (mustache and eyebrows), hair length, and curl. To locate the positions of the genes, the researchers used genome wide association studies (GWAS) with differing datasets. The datasets used were the dachshund dataset, CanMap dataset, and the Portuguese water dog (PWD) dataset. The dachshund dataset contained 96 dachshunds that had one of three coat phenotypes: wire-haired with furnishings, long-haired, and smooth hair. The CanMap dataset consisted of a broad range of phenotypes, and it included 903 dogs from 80 different breeds. The Portuguese water dog (PWD) dataset contained 76 PWDs and the dogs either had curl or did not. To determine the genetic basis for each trait the same general method was used. First, a GWAS was performed within a breed's dataset (PWD dataset or dachshund dataset) to determine the associated locus. This was followed by another GWAS of the CanMap dataset to double the check the associated locus determined in step one. Using the CanMap dataset ruled out potential false positives that may have shown up within the breed's dataset. The CanMap data set was set up into two groups: cases and controls. The control was determined to be the phenotype that was absent (i.e. no furnishings). Following this, fine-mapping of the associated haplotype was performed to refine it. The refined haplotype was then sequenced to determine possible mutations.
Three genes were found to be responsible for the dog pelage variations, and these genese were RSPO2, KRT71, and FGF5. RSPO2 controlled the presence or absence of furnishings, and it was determined to be a plausible candidate for the this trait, because the gene's product is involved in the pathway that establishes hair follicles. An insertion of 167 bp in the 3' untranslated region was associated with the presence of furnishings. Since the mutation of this gene was in the untranslated region, the researchers wanted to examine if the mutation resulted in a change of expression levels. A change was detected, and the change was determined to be a three-fold increase in the transcript of the RSPO2 gene. FGF5 was determined to be one of the genes responsible for pelage length. This gene was determined to have a mutation that changed a cysteine to a phenylalanine, and it resulted in the phenotype of longer hair. The gene KRT71 was identified to be responsible for curly pelages. In this gene, an SNP was located, and it resulted in the coding of a tryptophan rather than an arginine.
Summary:
This paper provides convincing evidence that supports the researchers's belief that they have located three genes that are responsible for variation in dog coat. The figures that the researchers provide are persuasive, and I believe that they were extremely helpful in assisting the reader understand the methods performed and the results obtained. Surprisingly, the most simple figure, figure 3, was the pictorial that provided the summary of the entire results, and aided in tying together a few loose ends. It is important to keep these results in perspective, however, because the three isolated genes do not explain all of the pelage phenotypes within dog breeds. The researchers mention that these three gene combinations are only seen in the phenotypes of 95% of the sampled dogs. Thus, there are more genes within the dog genome that can affect pelage phenotype. Most importantly, this paper demonstrated what is stressed to us most days in class; that one gene, one function, one phenotype is probably the exception to the rule, not the rule. This paper capitalized on this idea, as the researchers found that three genes primarily controlled all the different phenotypes of dog pelage (3 genes were responsible for 7 different dog pelage phenotypes). Additionally, while it is not mentioned, the approach used in this paper seems to demonstrate the value of the genome wide association studies. By using the methods outlined in the paper there seems to be real medical significance. If only three genes can create the broad variation responsible for dog coat, then such studies may provide insight into the variation among human diseases and certain populations.
Figures:
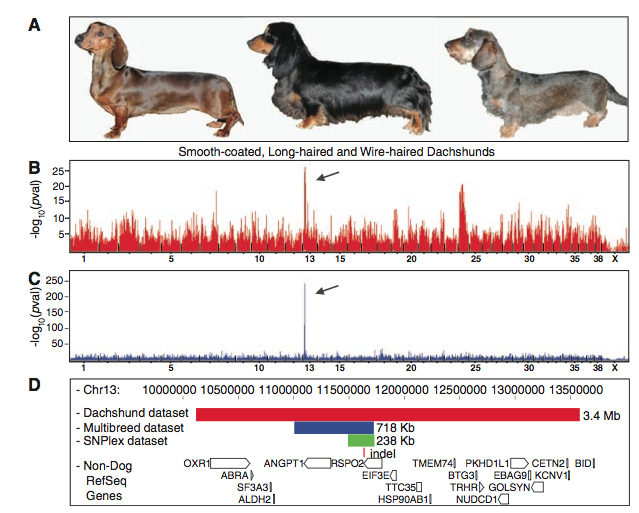
Figure 1: This diagram shows how the gene, RSPO2, was identified to be the gene associated with furnishings. (A) This diagram depicts the three coat phenotypes within the dachshund breed: smooth on the left, long-haired in the middle, and wire-haired (furnishings) on the right. It is provided to give a more concrete idea of what the researchers are referring to when they mention coat phenotypes. (B) This figure indicates the genome wide assessment (GWAS) results for the dachshund dataset. The wire-haired dogs were used as cases, and the long-coat and smooth-coat were the controls. The x-axis represents the entire genome with a tick mark below the x-axis to represent a chromosome. The y-axis is the –log10 of the p-values. A higher –log10 P-value indicates that the traits are closely linked. The arrow indicates the loci where the best p-value was found (3.35 x 10-27). This loci is on chromosome 13. The other raised vertical bars represent other p-values, however they are false-positives. (C) This figure is another set of results from a genome wide assessment (GWAS). However, unlike panel B, this GWAS was performed with the CanMap dataset (903 dogs from 80 breeds). This GWAS was performed to rule out the false-positives and confirm the association found in figure 1B. The x-axis represents the entire genome with a tick mark below the x-axis to represent a chromosome. The y-axis is the –log10 of the p-values. Again, a higher –log10 P-value indicates that the traits are closely linked. This linkage analysis confirmed the results of panel 1B (D) This figure portrays the regions identified from the genome wide associations of the dachshund breed dataset and the CanMap dataset that have been refined through fine-mapping. The red bar represents the dachshund's haplotype (3.4 Mb), the blue bar indicates the haplotype (718 Kb) found within the CanMap, and the green bar depicts the haplotype (238 Kb) that has been refined through fine-mapping. The red vertical line underneath the haplotypes represents an indel (insertion-deletion) that is located within all three haplotypes. The pointed rectangles, underneath the bars and deletion site, represent genes that are found in this particular region of chromosome 13. The way the arrows point on the white, pointed rectangles (genes) represents the direction of the reading frame. The gene RSPO2 is located within all three datasets, and the indel appears to be located within this gene.

Figure 2: This figure depicts regions within the datasets that distinguish two genes that are each responsible for the length and curl of the dog coat. (A) This figure is a genetic map of a particular region on chromosome 32 that is associated with pelage length. The red bar indicates the 520 Kb region found within the dachshund dataset, the blue bar represents the 125 Kb region found within the CanMap dataset, and the green bar represents the refined 67 Kb region after fine mapping. The single nucleotide polymorphism (SNP) that was most highly associated within the three regions (red, blue, green bars) is indicated by the red vertical bar underneath the 67 Kb region. The white, pointed rectangles indicate genes within the region of chromosome 32 being analyzed. The associated SNP appears to be within gene FGF5. The way the arrows point on the white, pointed rectangles (genes) represents the direction of the reading frame. (B) This picture depicts the trait that is being sought– pelage curl. The depictions show two varieties of pelage within the Portuguese water dog, curly (two left pictures), and wavy (two right pictures). (C) This figure is a fine mapping of the region believed to be associated with pelage curl. The green bar represents the 32 Kb haplotype that was located through fine mapping. This region, as indicated by the open, pointed rectangular boxes below the green line, contains two KRT genes. The SNP that results in a curly haired phenotype is located on gene KRT71. The SNP positions are indicated by vertical red bars underneath the green bar. Two SNPs are indicated in this figure, however, after sequencing the best SNP was located. It is the second SNP under the green bar. Again, the way the arrows point on the white, pointed rectangles (genes) represents the direction of the reading frame.
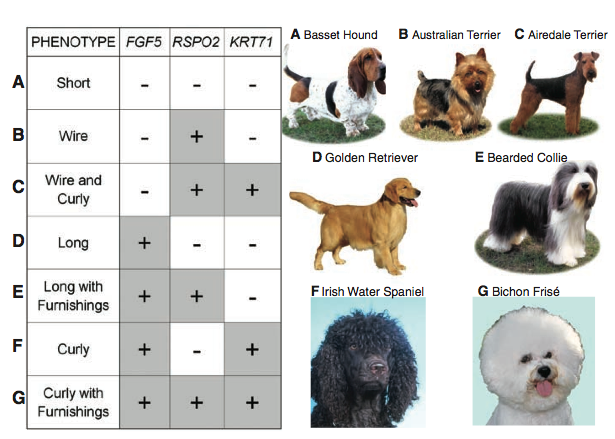
Figure 3: Seven different dog pelage phenotypes are a result of the presence/absence of the variant alleles. The three genes found to be responsible for dog coat variation are listed across the table (FGF5, RSPO2, and KRT71). A (+) sign indicates the presence of the variant allele (i.e. furnishings), and a (-) sign indicates the variant alleles absence (presence of the ancestral allele). The dog pictures on the right are corresponding pictorials that depict the phenotype caused by the combination of the three variant alleles. (A) Short hair phenotypes, as seen in the Bassett Hound, are the result of no variant alleles. (B) The wire-haired trait as seen in the Australian terrier is the result of one variant allele, RSPO2 . (C) The curly, wire-haired phenotype (Airedale Terrier) is the result of two variant alleles, RSPO2 and KRT71. (D) The long-haired phenotype is the result of the variant allele FGF5, and this phenotype is seen in golden retrievers. (E) Long pelages with furnishings, are the result of the variant alleles in FGF5 and RSPO2. This phenotype is seen in Bearded Collies. (F) Curly coats, as seen in the Irish Water Spaniel are the result of two variant alleles FGF5 and KRT71. (G) A curly pelage with phenotypes is the result of all three variant alleles. This phenotype is seen within the Bichon Frisé.
References:
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, VonHoldt BM, Rhue A, Boyko A, Byers A, et. al. Coat variation in the domestic dog is governed by variants in three genes. Science. 2009; 236:150-153.