Review of "Coat Variation in the Domestic Dog is Governed by Variants in Three Genes"
Introduction
The researchers set out to figure out what causes variations in fur phenotypes in dogs using genomic approaches. They broke the different variations into having or not having different combinations of wire-hair/furnishings, long fur, and curly fur. Three sets of dogs were used as the source of the data for the experiments: (i) 96 dachshunds which had dogs with wire-hair with furnishings (the mustache and eyebrows some dogs have), smooth short hair, and smooth long hair; (ii) 76 Portuguese water dogs that either did or did not have curly fur; (iii) 903 dogs from 80 breeds with a wide array of phenotypes (this data set was termed CanMap).
Method
All three fur traits were mapped using the same general method:
(1) A genome-wide association study (GWAS) was done within the breed data set that displayed the trait being mapped to find the locus most closely associated with the trait.
(2) A second genome-wide association study was done with the CanMap data set to again find loci closely associated with the trait in question.
(3) Fine-mapping within the closely associated regions further confined the area being investigated as being specific to that trait.
(4) This constrained region was then sequenced to find specific mutations that could be the cause of the trait.
Results and Dicussion
Wire-hair & Furnishings
Comparison of the results of the two GWASs found a 718-kb area of variation associated wire-hair and furnishings. The fine-mapping of this region further cut down the common region to 238-kb, within which only one gene was included: the R-spondin-2 (RSPO2) gene. RSPO2 is a good candidate because it is involved in a pathway for the establishment of hair follicles, and has also been tied to a specific type of tumor most often found in dogs with furnishings. Sequencing of RSPO2 from 7 breeds found a 167 base pair insertion in the 3' untranslated region (UTR) of RSPO2 in breeds with furnishings. The conclusion that the insertion mutation is responsible for a dog having furnishings was supported by the fact that 297 of the 298 dogs with furnishings were either homozygous or heterozygous for the insertion and all 406 dogs without furnishings were homozygous for the unmutated version of RSPO2. This alignment also suggests a dominant mode of inheritance for the trait, which would be in line with the proposed molecular effect of the mutation: an increase in RSPO2 transcripts, an effect common for elements in the 3' UTR and supported by analysis of tissue biopsies from furnished dogs. An increase in transcripts would result in increased translation, and having more than a normal level of protein is common in dominant disorders or mutations.
Hair Length
Both the GWAS of the the dachshunds data set and the GWAS of the CanMap data set found regions of high association that included the FGF5 gene, which had been implicated in connection to hair length in previous studies of dogs, cats, and mice. Fine-mapping of these regions cut the region down to 67-kb, but still included the FGF5 gene. The single nucleotide polymorphism (SNP) with the highest association, even after the region was sequenced, was in the first exon of FGF5 and changed a cysteine to a phenylalanine in the protein product of the gene. The mode of inheritance for the the trait appears to recessive as all long-haired dogs were homozygous for the SNP, while short or wire-haired dogs could be either heterozygous or homozygous for not having the SNP. This mutation is the only cause of long hair in dogs, as 3 breeds with long hair did not have the mutation or show any association to the area near the gene.
Curly Coat
Both a GWAS within the Portuguese water dog population and a GWAS within the CanMap found SNPs close to each other on chromosome 27. Fine-mapping of the region between the two SNPs found that the area included two keratin genes. Sequencing of most of the region found a specific SNP in the KRT71 keratin gene that was very closely associated with the trait. The SNP occurs in the protein-encoding part of the gene and changes an arginine to a tryptophan. This alteration could affect cellular targeting, receptor binding, or proper folding of the protein. The conclusion that this mutation is a cause of curly hair is supported by previous experiements, which have described mutations in KRT71 in curly haired mice.
Combined
Various combinations of these three mutations were found to account for the fur phenotypes of 95% of the dogs tested, which included 108 breeds out of the approximately 160 recognized by the American Kennel Club. They typed 622 dogs for the 3 mutations they found, and the mutations explained the phenotype for each dog:
None of the 3 mutations: short-haired breeds
RSPO2 mutation only: wire-hair and furnishings
RSPO2 and KRT71 mutations: curled or kinked wire-hair
RSPO2 and FGF5 mutations: long soft coats with furnishings
FGF5 mutation only: long, straight hair
FGF5 and KRT71 mutations: long and curly coats
All 3 mutations: long and curly with furnishings
The KRT71 mutation was not found by itself in any curly haired dogs. The authors' reasoning behind this was that the hair had to be of a certain length to curl. None of the three mutations isolated in the experiments were found when gray wolves were typed for them, suggesting that they are not the ancestral versions of the genes. The high degree of similarity in the areas around the mutations suggests that each mutation occured once and then was transferred to other breeds recently, likely due to artificial selection by humans.
Figures
Figure 1
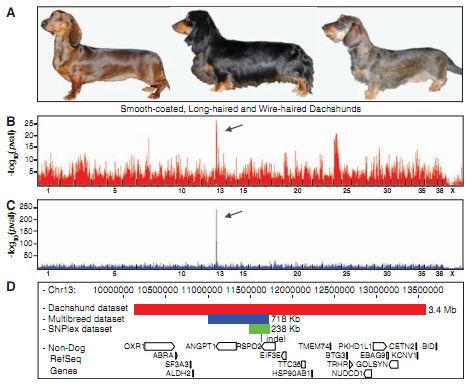
Figure 1A- This is just a pictorial representation of dachshunds exhibited the fur phenotypes that the authors used the population to investigate: smooth-coat on the left, long hair in the middle, and wire-hair with furnishings on the right. This figure just serves as a visualization for the reader of what the authors are referring to when discussing long, short, and wire-hair.
Figure 1B- This is a graphical representation of the GWAS performed within the dachshunds population in order to find regions associated with wire-hair. The x-axis is the genome, with each gap between hashes being one chromosome, and the y-axis is the negative log of the P value. The P value is the chance that the variation and the trait are associated purely by chance, so the authors were looking for what locations had the smallest P values, the smallest chance that any association observed was simply due to chance. The negative log of a small value is large, so the highest peaks are places of the lowest chance that the observed connection between variation and trait is due to random chance.
Figure 1C- This figure is similar to 1B, the only difference is that the data set used to generate this GWAS was the CanMap data set. The y-axis scale is also 10x the scale on the 1B, showing that this association is more strongly coorelated with wire-hair, likely an effect of having almost 10x as many dogs in the CanMap data set as in the dachshunds data set.
Figure 1D- This is a representation of each step they took to limit down the section that needed to be sequenced, and also what genes are found in the shared regions. The red rectangle is the associated region found with the dachshunds data set, the blue rectangle is that could with the CanMap data set, and the green rectangle is the result of fine-mapping between the two to find an area of homozygosity between the two. Below the green rectangle the insertion found through sequencing and its location is shown. The open boxes on the bottom of the figure show genes that are in the regions covered by any of the rectangles. We can see that by the time the region is limited all the way down after fine-mapping, only the RSPO2 gene is within the region.
Figure 2
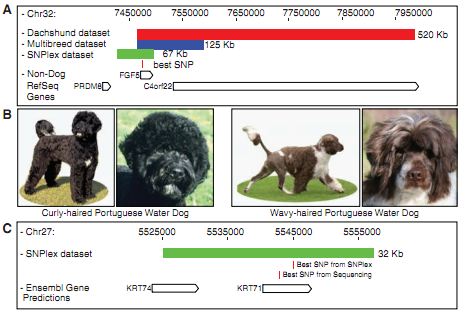
Figure 2A- The layout of this figure resembles that of 1D because it is showing the same things, just for hair length instead of wire-hair. The color coding for rectangles is the same as 1D: red is from dachshunds GWAS, blue is from CanMap GWAS, and the green is the result of fine-mapping between the two. The most strongly associated SNP is shown in it's position within the region and gene, and the open boxes again show genes in the regions found to be related to the trait at various steps along the method.
Figure 2B- These are more pictures to give the reader an idea of exactly what fur trait, curly hair, is being investigated by the authors.
Figure 2C- This is supposed to be showing the same idea as 1D and 2A except for curly hair, but is cut down from those as it only shows the green rectangle of commonly shared area found after fine-mapping, the strongest associated SNP after fine-mapping, the strongest SNP association after sequencing, and the genes within the green rectangle. It shows that both stron SNPs found are within the KRT71 gene.
Figure 3
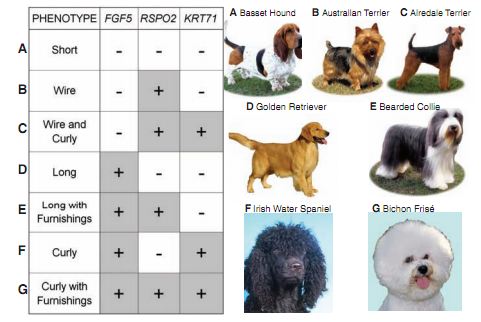
This figure is the summary figure for the paper. The table shows all the combinations of mutations found by the authors, and resultant phenotype labeled with a letter that cooresponds to a picture of a dog exhibiting that phenotype.
Opinion
The paper appeared to do a good job showing that the mutations they isolated are the causes of the traits they investigated. I would be very hesitant to say that these 3 mutations are the cause of all the fur phenotypic diversity in dogs, as they acknowledge that some dogs' phenotypes were not explained (specifically some long-haired dogs), and they only typed about 2/3 of the AKC breeds, which do not include other unrecognized breeds. I also wish that the GWAS data was included in the figures for each trait, instead of only for wire-hair. Though the authors make no attempts no extrapolate beyond dog hair phenotypes, some people might try to use the paper to say something about humans, which would be risky at best because a lot of the ability for a single mutation to be associated with a trait stems from the high level of past artificial selection in dogs, a feature that has not occured in humans. Overall this was a good paper that I found very interesting.
Reference
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, VonHoldt BM, Rhue A, Boyko A, Byers A, et. al. Coat variation in the domestic dog is governed by variants in three genes. Science. 2009; 236:150-153.
The researchers set out to figure out what causes variations in fur phenotypes in dogs using genomic approaches. They broke the different variations into having or not having different combinations of wire-hair/furnishings, long fur, and curly fur. Three sets of dogs were used as the source of the data for the experiments: (i) 96 dachshunds which had dogs with wire-hair with furnishings (the mustache and eyebrows some dogs have), smooth short hair, and smooth long hair; (ii) 76 Portuguese water dogs that either did or did not have curly fur; (iii) 903 dogs from 80 breeds with a wide array of phenotypes (this data set was termed CanMap).
Method
All three fur traits were mapped using the same general method:
(1) A genome-wide association study (GWAS) was done within the breed data set that displayed the trait being mapped to find the locus most closely associated with the trait.
(2) A second genome-wide association study was done with the CanMap data set to again find loci closely associated with the trait in question.
(3) Fine-mapping within the closely associated regions further confined the area being investigated as being specific to that trait.
(4) This constrained region was then sequenced to find specific mutations that could be the cause of the trait.
Results and Dicussion
Wire-hair & Furnishings
Comparison of the results of the two GWASs found a 718-kb area of variation associated wire-hair and furnishings. The fine-mapping of this region further cut down the common region to 238-kb, within which only one gene was included: the R-spondin-2 (RSPO2) gene. RSPO2 is a good candidate because it is involved in a pathway for the establishment of hair follicles, and has also been tied to a specific type of tumor most often found in dogs with furnishings. Sequencing of RSPO2 from 7 breeds found a 167 base pair insertion in the 3' untranslated region (UTR) of RSPO2 in breeds with furnishings. The conclusion that the insertion mutation is responsible for a dog having furnishings was supported by the fact that 297 of the 298 dogs with furnishings were either homozygous or heterozygous for the insertion and all 406 dogs without furnishings were homozygous for the unmutated version of RSPO2. This alignment also suggests a dominant mode of inheritance for the trait, which would be in line with the proposed molecular effect of the mutation: an increase in RSPO2 transcripts, an effect common for elements in the 3' UTR and supported by analysis of tissue biopsies from furnished dogs. An increase in transcripts would result in increased translation, and having more than a normal level of protein is common in dominant disorders or mutations.
Hair Length
Both the GWAS of the the dachshunds data set and the GWAS of the CanMap data set found regions of high association that included the FGF5 gene, which had been implicated in connection to hair length in previous studies of dogs, cats, and mice. Fine-mapping of these regions cut the region down to 67-kb, but still included the FGF5 gene. The single nucleotide polymorphism (SNP) with the highest association, even after the region was sequenced, was in the first exon of FGF5 and changed a cysteine to a phenylalanine in the protein product of the gene. The mode of inheritance for the the trait appears to recessive as all long-haired dogs were homozygous for the SNP, while short or wire-haired dogs could be either heterozygous or homozygous for not having the SNP. This mutation is the only cause of long hair in dogs, as 3 breeds with long hair did not have the mutation or show any association to the area near the gene.
Curly Coat
Both a GWAS within the Portuguese water dog population and a GWAS within the CanMap found SNPs close to each other on chromosome 27. Fine-mapping of the region between the two SNPs found that the area included two keratin genes. Sequencing of most of the region found a specific SNP in the KRT71 keratin gene that was very closely associated with the trait. The SNP occurs in the protein-encoding part of the gene and changes an arginine to a tryptophan. This alteration could affect cellular targeting, receptor binding, or proper folding of the protein. The conclusion that this mutation is a cause of curly hair is supported by previous experiements, which have described mutations in KRT71 in curly haired mice.
Combined
Various combinations of these three mutations were found to account for the fur phenotypes of 95% of the dogs tested, which included 108 breeds out of the approximately 160 recognized by the American Kennel Club. They typed 622 dogs for the 3 mutations they found, and the mutations explained the phenotype for each dog:
None of the 3 mutations: short-haired breeds
RSPO2 mutation only: wire-hair and furnishings
RSPO2 and KRT71 mutations: curled or kinked wire-hair
RSPO2 and FGF5 mutations: long soft coats with furnishings
FGF5 mutation only: long, straight hair
FGF5 and KRT71 mutations: long and curly coats
All 3 mutations: long and curly with furnishings
The KRT71 mutation was not found by itself in any curly haired dogs. The authors' reasoning behind this was that the hair had to be of a certain length to curl. None of the three mutations isolated in the experiments were found when gray wolves were typed for them, suggesting that they are not the ancestral versions of the genes. The high degree of similarity in the areas around the mutations suggests that each mutation occured once and then was transferred to other breeds recently, likely due to artificial selection by humans.
Figures
Figure 1
Figure 1A- This is just a pictorial representation of dachshunds exhibited the fur phenotypes that the authors used the population to investigate: smooth-coat on the left, long hair in the middle, and wire-hair with furnishings on the right. This figure just serves as a visualization for the reader of what the authors are referring to when discussing long, short, and wire-hair.
Figure 1B- This is a graphical representation of the GWAS performed within the dachshunds population in order to find regions associated with wire-hair. The x-axis is the genome, with each gap between hashes being one chromosome, and the y-axis is the negative log of the P value. The P value is the chance that the variation and the trait are associated purely by chance, so the authors were looking for what locations had the smallest P values, the smallest chance that any association observed was simply due to chance. The negative log of a small value is large, so the highest peaks are places of the lowest chance that the observed connection between variation and trait is due to random chance.
Figure 1C- This figure is similar to 1B, the only difference is that the data set used to generate this GWAS was the CanMap data set. The y-axis scale is also 10x the scale on the 1B, showing that this association is more strongly coorelated with wire-hair, likely an effect of having almost 10x as many dogs in the CanMap data set as in the dachshunds data set.
Figure 1D- This is a representation of each step they took to limit down the section that needed to be sequenced, and also what genes are found in the shared regions. The red rectangle is the associated region found with the dachshunds data set, the blue rectangle is that could with the CanMap data set, and the green rectangle is the result of fine-mapping between the two to find an area of homozygosity between the two. Below the green rectangle the insertion found through sequencing and its location is shown. The open boxes on the bottom of the figure show genes that are in the regions covered by any of the rectangles. We can see that by the time the region is limited all the way down after fine-mapping, only the RSPO2 gene is within the region.
Figure 2
Figure 2A- The layout of this figure resembles that of 1D because it is showing the same things, just for hair length instead of wire-hair. The color coding for rectangles is the same as 1D: red is from dachshunds GWAS, blue is from CanMap GWAS, and the green is the result of fine-mapping between the two. The most strongly associated SNP is shown in it's position within the region and gene, and the open boxes again show genes in the regions found to be related to the trait at various steps along the method.
Figure 2B- These are more pictures to give the reader an idea of exactly what fur trait, curly hair, is being investigated by the authors.
Figure 2C- This is supposed to be showing the same idea as 1D and 2A except for curly hair, but is cut down from those as it only shows the green rectangle of commonly shared area found after fine-mapping, the strongest associated SNP after fine-mapping, the strongest SNP association after sequencing, and the genes within the green rectangle. It shows that both stron SNPs found are within the KRT71 gene.
Figure 3
This figure is the summary figure for the paper. The table shows all the combinations of mutations found by the authors, and resultant phenotype labeled with a letter that cooresponds to a picture of a dog exhibiting that phenotype.
Opinion
The paper appeared to do a good job showing that the mutations they isolated are the causes of the traits they investigated. I would be very hesitant to say that these 3 mutations are the cause of all the fur phenotypic diversity in dogs, as they acknowledge that some dogs' phenotypes were not explained (specifically some long-haired dogs), and they only typed about 2/3 of the AKC breeds, which do not include other unrecognized breeds. I also wish that the GWAS data was included in the figures for each trait, instead of only for wire-hair. Though the authors make no attempts no extrapolate beyond dog hair phenotypes, some people might try to use the paper to say something about humans, which would be risky at best because a lot of the ability for a single mutation to be associated with a trait stems from the high level of past artificial selection in dogs, a feature that has not occured in humans. Overall this was a good paper that I found very interesting.
Reference
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, VonHoldt BM, Rhue A, Boyko A, Byers A, et. al. Coat variation in the domestic dog is governed by variants in three genes. Science. 2009; 236:150-153.