This web page was produced as an assignment for an undergraduate course at Davidson College.
How Well Does the Popular Press Report on new Scientific Discoveries?
On May 17, 2012 two seperate articles were published online in Science that gave further support to the idea that science has only scratched the surface of the complexity of our genomes. With the debunking of the "one disease, one variant" theory by discoveries of the complex genetic factors behind development of genetic diseases like muscular dystrophy, many scientists had hypothesized a "common disease, common variant" theory. That is, that a disease that is common in a population is caused by the same common variation, or mutation, in that a person's DNA. The two studies from Science, however, provide strong evidence against this theory as well by showing that a high number of very rare genetic mutations may be one of the key contributors to the development of many common diseases. The two projects, one from the University of Washington in Seattle, and the other from GlaxoSmithKline, looked at different parts of people's genomes, but saw highly similar results leading to the shared conclusion.
So what are the consequences of the “common disease, common variant” theory being disproven? For one thing, in today’s day and age when the numerous values of a fully sequenced human genome for $1000 are being touted left and right, the results from these studies are shedding some serious doubt on the utility of a sequenced genome to a doctor. If most diseases are at least heavily influenced by very rare small genetic mutations, it would be very hard to identify if a person was susceptible to a certain disease, simply because the rare mutation they have has not been discovered yet. Also compounding the problem is that with some of the mutations being a single base pair, it is sometimes difficult to determine if what seems to be a mutation is really what it appears or simply a mistake in the sequencing. And even if a mutation is identified, it is extremely difficult to identify the effect of the mutation without a large sample size to compare it to. The fact that most of the mutations were population specific also means that comparing relationships between certain mutations and diseases across populations is more difficult.
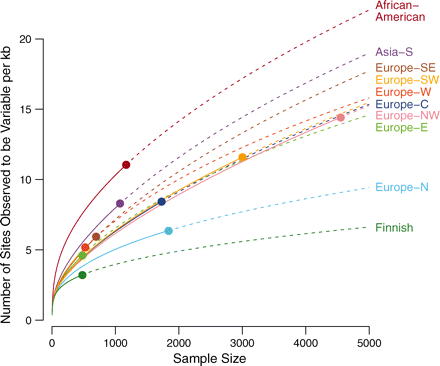 Number of variants per kilobase of DNA in various populations. Observed values are marked with dots. Solid lines indicate hypergeometric expectations and dashed lines indicate jackknife projections. Different populations have varying amounts of SNVs.
Number of variants per kilobase of DNA in various populations. Observed values are marked with dots. Solid lines indicate hypergeometric expectations and dashed lines indicate jackknife projections. Different populations have varying amounts of SNVs.
University of Washington in Seattle Study
In the University of Washington in Seattle study, the researchers sampled 15,585 human protein-coding genes from a population of 2440 individuals of both European and African ancestry. They detected of 500,000 single-nucleotide variants (SNVs) with 86% being rare, 82% previously unknown, and 82% population specific. Also, almost 96% of the SNVs that were predicted to be functionally important were rare. Finally, they determined that this large number of exceedingly rare functional variants is due to explosive, recent (in evolutionary terms) population growth combined with weak purifying selection (Tennessen et al. 2012).
GlaxoSmithKline Study
In the study conducted by GlaxoSmithKline the researchers sequenced 202 genes in 14,002 individuals that encode for drug targets. They found that there was a rare variant every 17 bases, with 95% of the SNVs being rare and 90% of them being previously unknown. They also concluded that these variants were due to enormous recent population growth and weak purifying selection, and that many of the variants they found were deleterious and related to disease risk (Nelson et al. 2012).
How are the findings reported in the popular press?
The results of the studies were reported in the same story in the New York Times when they were released. The NYT report does an extremely good job of being an intermediary between the detailed scientific jargon of the Science articles and the general public. The scientific articles are filled with statistical tests to determine if the data that they saw was significant or not, and the NYT article does a good job of filtering this out to only the conclusions, which can be understood by the general public. It isn’t very necessary for the general public to know what kind of statistical test the scientists used to claim that their results were significant, as this is too detailed and specialized for the average person to understand from a small article. It sets the results of the studies in context by talking about past genetic theories on disease (i.e. “common disease, common variant”), the relationship to and effects on personal genomics and personalized medicine from these results, and briefly talking about the reason this research is happening now (new DNA sequencing technology has made sequencing faster and more accurate). They also talk about a scientist unrelated to the two studies to show that the ideas put forth in the studies have significance and supporters in the rest of the scientific world. The NYT article also briefly touches on the thing that makes not only the results of these studies exciting, but their methods as well, by mentioning that they are particularly extensive surveys of genes. The author of the NYT article also does a good job of not using the superlative or exaggerated statements that are so often a hallmark of journalism, and that so strongly clash with the measured, cautious mindset of science. The author avoids another common feature of scientific journalism that Kua et al. mention in that by simplifying the scientific jargon of the journal articles for the general public, Wade (the author of the NYT article) does not change the core information being portrayed. Wade assumes that the reader knows some background information (the idea of a human’s sequenced genome being used for medical purposes, basic knowledge of what DNA is, etc), but provides a solid framing for the results of the studies in the context of today’s scientific and medical world, leading the reader through common connections between this new data and other ideas. (Wade 2012).
In a watchdog role, Wade also performs admirably. He brings up the medical implications of the new data, as well as the place of the data in the bigger picture. He fails to bring up any social or ethical implications of the new studies, but this is more due to the nature of the data than any fault of Wade’s. There is a racial aspect to these studies, with most of the data split into that for populations of European and African descent, but the data supports this division as the SNVs the researchers saw were mostly population specific. In talking about how the new data casts some doubt on the idea of “quick medical payoffs from the human genome project”, Wade makes the reader think about this idea and the role that personal genomics will come to play in medicine (Wade 2012). He also manages to avoid hyperbole by bringing up the uses that a fully sequenced patient genome would have in medicine, even if it isn’t the magic bullet that is has recently been touted as.
Finally, Wade gives readers the tools needed to analyze the evidence and issues themselves to a certain degree. As mentioned previously, he gives a very good description of the place that the new evidence has in the current medical and scientific field. He doesn’t give any information as to directions of future research, but instead brings up the flaws of our current understanding of the topic, allowing the reader to think about future research topics on their own.
Some scientists may claim that Wade does not go into enough detail with his description of the science and data behind the new evidence from the new studies, but how much detail is really necessary? A journalist should give the reader the tools needed to analyze the information given, and should translate the scientific vocabulary into something more manageable for the average reader, but can one have too many tools? Or what happens if it is a bad translation? What level of detail is necessary for the general public to understand new scientific data? Especially in papers like the two Science articles, which are filled with statistics and parameters and tests just to allow the researchers to say that their data is significant, the general public probably does not have to know anything but the conclusions. If they are curious as to the specifics, a link is provided to each journal article. Wade manages to distill pages and pages of dense statistical tests down to a very manageable article that gives the general public a very good idea of what the new data from the studies means and how it fits into the bigger picture, even if he doesn’t fully explain the complicated mechanisms behind the results because his audience does not need to know them.
New York Times story
Nature Article 1: Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes
Nature Article 2: An Abundance of Rare Functional Variants in 202 Drug Target Genes Sequenced in 14,002 People
Sources
Eunice Kua, Michael Reder, and Martha J. Grossel. 2004. Science in the News: A Study of Reporting Genomics. Public Understanding of Science. 13: 309–322.
Nelson, Matthew R., Daniel Wegmann, Margaret G. Ehm, Darren Kessner, Pamela St. Jean, Claudio Verzilli, Judong Shen, Zhengzheng Tang, Silviu-Alin Bacanu, Dana Fraser, Liling Warren, Jennifer Aponte, Matthew Zawistowski, Xiao Liu, Hao Zhang, Yong Zhang, Jun Li, Yun Li, Li Li, Peter Woollard, Simon Topp, Matthew D. Hall, Keith Nangle, Jun Wang, Gon Alo Abecasis, Lon R. Cardon, Sebastion Z. Ilner, John C. Whittaker, Stephanie L. Chissoe, John Novembre, and Vincent Mooser. "An Abundance of Rare Functional Variants in 202 Drug Target Genes Sequenced in 14,002 People." Science 337 (2012): 100-04. Print.
Tennessen, Jacob A., Abigail W. Bigham, Timothy D. O'Connor, Wenqing Fu, Eimear E. Kenny, Simon Gravel, Sean McGee, Ron Do, Xiaoming Liu, Goo Jun, Hyun Min Kang, Daniel Jordan, Suzanne M. Leal, Stacey Gabriel, Mark J. Rieder, Goncalo Abecasis, David Altshuler, Deborah A. Nickerson, Eric Boerwinkle, Shamil Shunyaev, Carlos D. Bustamante, Michael J. Bamshad, and Joshua M. Akey. "Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes." Science 337 (2012): 64-69. Print.
Wade, Nicholas. "Rare Genetic Mutations May Underpin Diseases." The New York Times. The New York Times, 18 May 2012. Web. 03 Feb. 2013.
Genomics Page
Biology Home Page
Ben Clarkson Home Page
Email Questions or Comments to beclarkson@davidson.edu.
© Copyright 2013 Department of Biology, Davidson College, Davidson, NC 28035