This web page was produced as an assignment for an undergraduate course at Davidson College.
Brett E. Johnson,1* Tali Mazor,1* Chibo Hong,1 Michael Barnes,2 Koki Aihara,3,4 Cory Y. McLean,1† Shaun D. Fouse,1 Shogo Yamamoto,3 Hiroki Ueda,3 Kenji Tatsuno,3 Saurabh Asthana,5,6 Llewellyn E. Jalbert,7 Sarah J. Nelson,7,8 Andrew W. Bollen,2 W. Clay Gustafson,9 Elise Charron,10 William A. Weiss,1,9,10 Ivan V. Smirnov,1 Jun S. Song,11,12 Adam B. Olshen,6,11 Soonmee Cha,1 Yongjun Zhao,13 Richard A. Moore,13 Andrew J. Mungall,13 Steven J. M. Jones,13 Martin Hirst,13 Marco A. Marra,13 Nobuhito Saito,4 Hiroyuki Aburatani,3 Akitake Mukasa,4 Mitchel S. Berger,1 Susan M. Chang,1 Barry S. Taylor,5,6,11‡ Joseph F. Costello1‡
1Department of Neurological Surgery, University of California, San Francisco, CA 94158, USA. 2Department of Pathology, University of California, San Francisco, CA 94158, USA. 3Genome Science Laboratory, Research Center for Advanced Science and Technology, University of Tokyo, Meguro-ku, Tokyo 153-8904, Japan. 4Department of Neurosurgery, University of Tokyo, Bunkyo-ku, Tokyo 113-8655, Japan. 5Department of Medicine, University of California, San Francisco, CA 94158, USA. 6Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, CA 94158, USA. 7Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, CA 94158, USA. 8Department of Radiology and Biomedical Imaging, University of California, San Francisco, CA 94158, USA. 9Department of Pediatrics, University of California, San Francisco, CA 94158, USA. 10Department of Neurology, University of California, San Francisco, CA 94158, USA. 11Department of Epidemiology and Biostatistics, University of California, San Francisco, CA 94158, USA. 12Institute for Human Genetics, University of California, San Francisco, CA 94158, USA. 13Michael Smith Genome Sciences Centre, British Columbia Cancer Agency, Vancouver, BC V5Z 4E6, Canada. *These authors contributed equally to this work. †Present address: 23andMe Inc., Mountain View, CA 94043, USA. ‡Corresponding author. E-mail: jcostello@cc.ucsf.edu ( J.F.C.); barry.taylor@ucsf.edu (B.S.T.)
John et al. sequenced and compared the mutations of sets of initial and recurrent gliomas in order to characterize their genetic relatedness and the patterns of their evolutionary relationship. Recurrent gliomas were found to share a large portion (54%) of their mutation profiles with initial tumors, which suggests that both tumor types developed from the same ancestral cancerous cell. Although no further evolutionary pattern (e.g. linear/branched) was found to be intrinsic to the recurrent gliomas, they did have one characteristic in common in that they were of a higher, more malignant tumor grade. By classifying almost all detectable somatic mutations (>98.7%) of TMZ-treated hypermutated recurrences as TMZ-associated, it was then determined that the drug introduced a unique selective pressure that contributed to the progressive malignancy of recurrences.
The scientific contribution of Johnson et al. to the field of clinical genomics is both substantial and exciting. The researchers present a proof-of-concept that 1) cancer recurrences should not be viewed as equivalent to the initial tumors and 2) current cancer therapies such as TMZ-treatment can induce the progression of malignancy in possible recurrences and should therefore be applied only upon the exhaustion of all other therapeutic options. These findings will hopefully serve to warn patients and doctors alike about the possible side effects of tumor treatments as well as to refocus the pharmaceutical research and development field into the direction of more specific medicine.
The research underlying these conclusions was presented in a succinct and impactful, but at times also unclear and possibly biased manner. Examples of the latter were:
I assume that the lack of answers to many of my questions might be attributable to the editorial restrictions of Science. If so, I would recommend that the editors consider revising their policy on brevity so that it does not force the reader to make assumptions for the truthfulness and reliability of the data shown by only relying on the editor’s judgment. Provided that there exist satisfying answers to my most important questions though, overall, I think this was a very educational paper.
Also, I love the fact that the research was supported by the Alex Lemonade Stand Foundation.
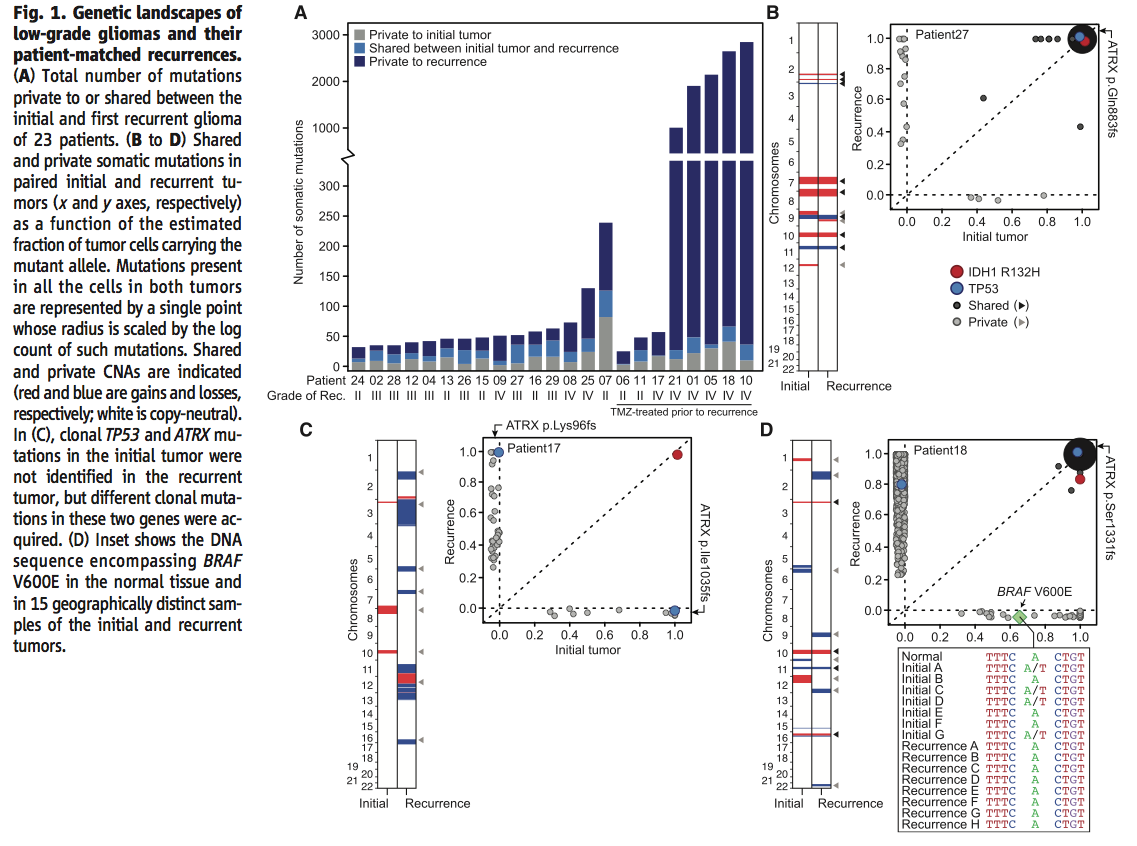
Figure 1 attempts to asses how genetically different recurrent gliomas are from the initial tumors developed by the respective patients. Johnson et al. compared the exomes of grade II tumors at initial diagnosis to those of their recurrences in a total of 23 cancer patients. The DNA was sequenced to an average 125-fold coverage, which allowed for about 10% mutation detection sensitivity.
Panel A demonstrates that recurrences share a large portion (54% average across all pairs) of the somatic mutations found on initial gliomas. This result supports a hypothesis that recurrences are genetically related to initial tumors. Importantly, the average appears to be considerably reduced by a set of TMZ-treated gliomas (patients 17, 21, 01, 05, 18 and 10) whose reccurences are of a higher grade (IV) and have accumulated a much larger number of private somatic mutations.
As can be inferred from the chromosomal map and the dot plot comparing initial and recurrent tumor mutation profiles (as a function of the estimated fraction of tumor cells carrying the mutant allele) in Panel B, the recurrent gliomas of some patients share significantly more mutations (>75 %) with the initial tumors. Those secondary gliomas were likely derived from initial tumors at a late stage of their evolution and provide evidence for linear clonal evolution.
Other patients’ recurrent gliomas, on the other hand, share significantly less mutations (<25%) with the initial tumors (Panel C). Those recurrent gliomas were likely derived from initial tumors at an early stage of their evolution and provide evidence for branched clonal evolution.
In order to evaluate the possibility of bias in the genetic relatedness data due to intratumoral heterogeneity and small sample sizes, the researchers examined DNA samples from geographically distinct regions of the tumors, focusing on individual mutations that were found to be private to either initial tumors or recurrences. Almost all mutations, represented by the BRAF V600E mutation specific to initial gliomas in Panel D, proved their specificity, so it can be safely concluded that no such bias existed.
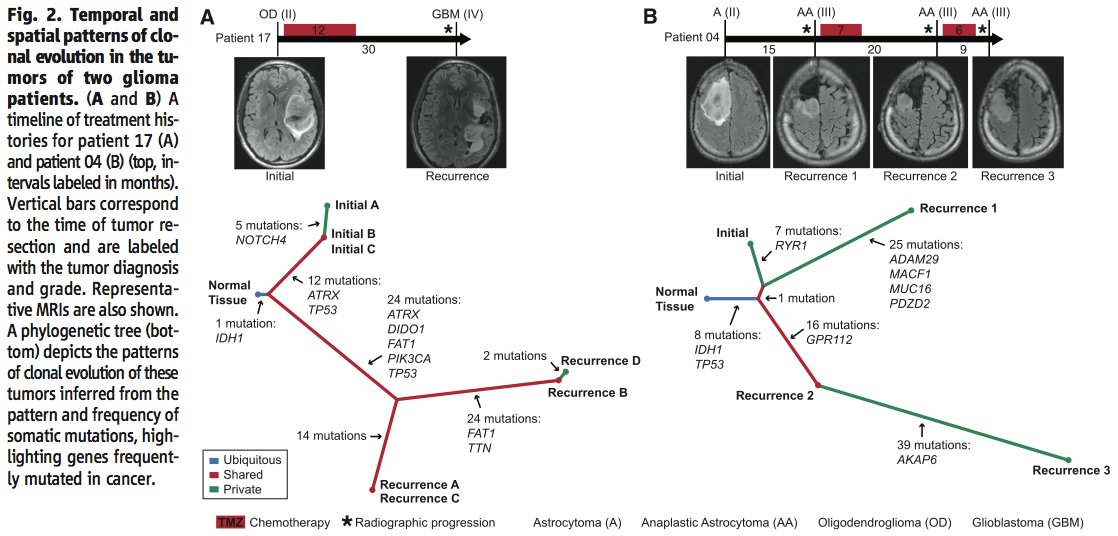
In order to analyze the pattern of tumor development from initial to recurrent, Johnson et al. were able to use the mutational profiles determined in Figure 1 to infer gliomal evolutionary relationships. As seen from the phylogenic tree in Panel A of Figure 2, three samples of initial glioma analyzed from Patient 17 show evidence of linear clonal evolution within the initial tumor (“Initial A” evolved from “Initial B” which evolved from “Initial C”) and have a common IDH1 mutant ancestor with four recurrent glioma samples. Nevertheless, even the mutations that were supposedly shared by initial and recurrent tumors, ATRX and TP53, proved to be of different origins. The overall branching pattern of the phylogenic tree therefore indicates a history of branched clonal evolution for the initial and recurrent gliomal cells.
Next, the researchers wanted to determine the pattern of evolution of sequential recurrences and whether it could be traced back to a single evolutionary stage in the initial glioma. Consequently, they sequenced the second and third tumor recurrences of Patient 04 and likewise inferred their phylogeny. As evident from the phylogenic tree in Panel B of the figure, although the initial glioma and all three recurrences share a common IDH1 and TP53 mutant ancestor, they did not diverge from it at a single evolutionary stage.Cells from the second recurrence appear to have branched off from the common ancestor during an earlier evolutionary time point than cells from the first one or from the initial tumor. Moreover, cells from the third recurrence appear to have evolved directly from the second recurrence. Hence, it can be concluded that there is no unequivocal pattern intrinsic to gliomal evolution.
Of note, the researchers were still able to identify a characteristic limited to almost all recurrences. As inferred from the timelines represented on top of both panels, recurrent gliomas were usually of a higher grade than the initial tumors.
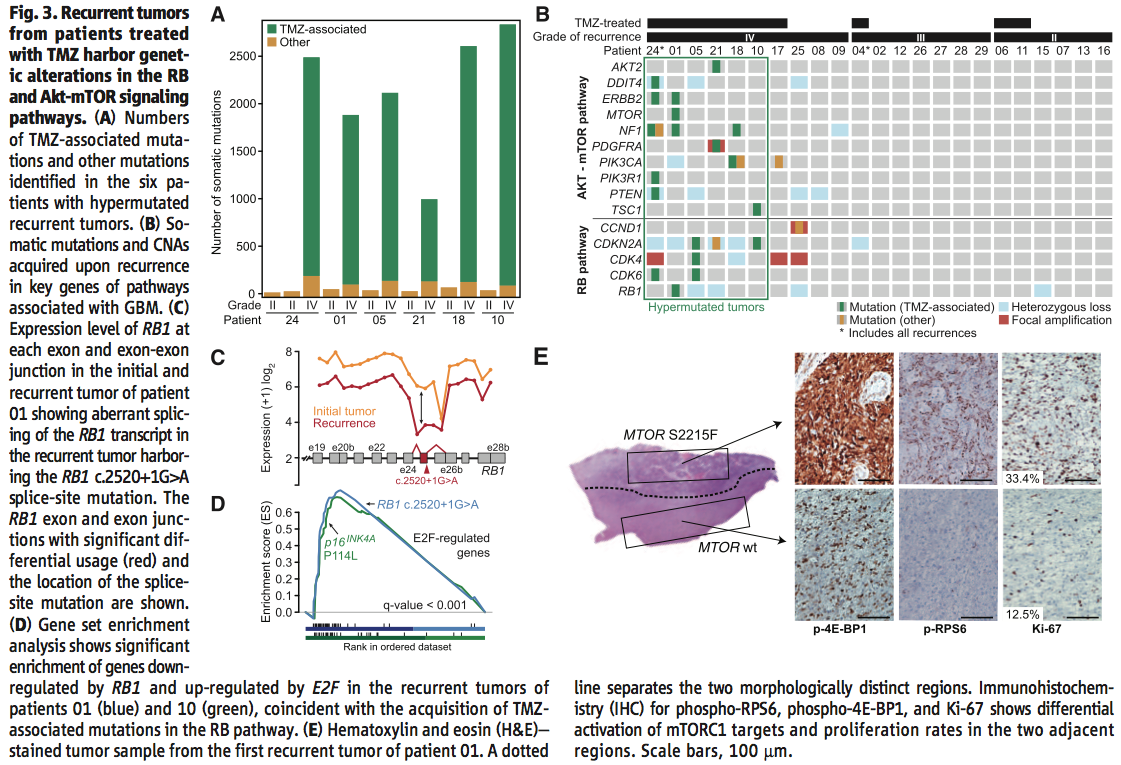
In the last figure of the paper, Johnson et al. investigate the effects of TMZ treatment on the mutational profiles of recurrent gliomas. For that purpose, the researchers classified a set of C>T/G>A recurrence-specific mutations in tumors subjected to TMZ treatment as “TMZ-associated.”
Panel A of the figure establishes that TMZ-associated somatic mutations comprise the better part (>98.7%) of the mutational profiles of tumor recurrences. TMZ is therefore a major mutation-inducing agent and likely contributes significantly to the malignant progression of initial gliomas to higher-grade recurrences.
In order to identify TMZ-associated mutations which likely contribute to higher recurrence malignancy, the researchers then focused on the RB and Akt-mTOR signaling pathways that were previously known to be associated with proliferation regulation. Panel B demonstrates that mutations in either of those pathways were present almost exclusively in Grade IV, TMZ-treated tumors, thereby validating their correlation to tumor malignancy.
Panel C compares the transcriptomes of initial and recurrent tumors for one such mutation, RB1. As per the lower RB1 mRNA concentration (y-axis coordinates) of the recurrent tumor, and as predicted by exome sequencing comparison (x-axis), the RB1c.2520+1G>A splice-site mutation appears to substantially lower RB1 expression overall.
Panel D, based on gene set enrichment score analysis, provides further evidence for disruption of RB1-mediated regulation caused by the RB1c.2520+1G>A mutation. Before the mutation, the p16INK4A and P114L genes are significantly enriched in an attempt to control the proliferation of cancer cells. At the occurrence of the mutation, however, the genes, which are normally regulated by RB1 and E2F antagonism, gradually stop being expressed and regulation is thereby imparied. Consequently, mutations in the RB pathway prove to be instrumental for facilitating cancer proliferation in recurrent gliomas.
Lastly, panel E presents the image results of an immunohistochemistry experiment for p-4E-BP1, p-RPS6 and Ki-67 (proliferation markers) in MTORwt (bottom row) and MTOR S2215F mutant (top row) tissue samples from a primary recurrence in Patient 1. Since MTOR is only mutant in one of the regions of the same tumor, comparison between the top and bottom rows of Panel E proves the existence of intratumoral heterogeneity. Moreover, the MTORS2215F region expresses higher levels of either protein sampled for and is therefore more active, thereby demonstrating that mutations in the AKT-mTOR pathway also serve to increase cancer malignancy.
Genomics Page
Biology Home Page
© Copyright 2014 Department of Biology, Davidson College, Davidson, NC 28035