This web page was produced as an assignment for an undergraduate
course at Davidson College.
Role of Superantigens in Staphylococcal and Streptococcal
Toxic Shock Syndrome
Overview
Toxic-shock syndrome (TSS) is a rapid-onset illness causing fever, hypotension,
rash, vomiting, diarrhea, and eventually multiple organ failure.
If not treated promptly, TSS is lethal (Bohach et al. 1990).
TSS is caused by the nonspecific stimulation of T lymphocytes by superantigens
that belong to a family of pyrogenic toxins produced by the bacteria Staphylococcus
aureus and Streptococcus pyogenes(Schlievert 1993). Infection
with S. aureus produces classical TSS, whereas S. pyogenes
causes a modified form of TSS known as either streptococcal TSS (Bohach
et al. 1990) or, more recently, toxic-shock-like syndrome (TSLS;
Schlievert 1993). TSLS displays many of the typical TSS symptoms
with the addition of severe soft tissue necrosis (Stevens 1995).
For simplicity, staphylococcal TSS henceforth will be referred to simply
as TSS, streptococcal TSS will be referred to as TSLS.
Staphylococcal TSS
S. aureusproduces two types of toxins that are implicated in
TSS: TSS toxin-1 (TSST-1) and staphylococcal exotoxins (SEs); the latter
occur in several serotypes, primarily B and C1. TSS-related S.
aureus infection can be broadly divided into menstrual (those involving
vaginal infection in menstruating women) and non-menstrual cases.
Ninety percent of menstrual cases are caused by TSST-1, non-menstrual cases
are evenly split between TSST-1 and SEs. SEs also play a role in
staphylococcal food poisoning (Bohach et al. 1990).
Menstrual TSS is usually related to tampon use; tampons increase the
vaginal concentration of oxygen, which stimulates TSST-1 production by
S.
aureus that normally reside in the vagina. High-absorbancy tampons
also sequester magnesium ions, which causes nutrient depletion in the vagina
and may simulate late log-phase conditions for resident S. aureus,
inducing TSST-1 secretion (Schlievert 1993). In the late 1970s and
early 1980s, a rise in incidence of menstrual TSS was related to use of
high-absorbancy tampons, such as the Rely
brand, that have since been taken off the market (Schlievert 1993, Bohach
et al. 1990).
The dependence of menstrual TSS on TSST-1 could be related to the ability
of TSST-1 (but not other pyrogenic toxins) to cross the vaginal mucosa.
Some epithelial cells are known to display TSST-1 receptors; these receptors
could be involved in endocytosis, transcytosis, and basolateral secretion
of TSST-1 by the vaginal epithelium (Bohach et al. 1990).
Non-menstrual onset of TSS is likely related to opportunistic S.
aureus infections in the vagina and elsewhere. A major class
of non-menstrual TSS is flu-associated; S. aureus can infect nasopharyngeal
tissues damaged by influenza infection and cause TSS by secreting TSST-1
or SEs (Schlievert 1993).
View Chime image of TSST-1
View Chime image of SE A
Get Chime
Streptococcal TSLS
TSLS was first described in 1987 (Bohach et al. 1990), as a result
it is not as well characterized as TSS. Group A streptococcal bacteria,
primarily S. pyogenes, cause TSLS by secreting streptococcal pyrogenic
exotoxins (SPEs) of three serotypes: A, B, and C. An additional toxin,
streptococcal superantigen (SSA) was recently discovered and may also be
involved, though its exact role is unknown. S. pyogenes infection
usually occurs via minor injuries or surgeries, though the exact portal
of infection is uknown for most TSLS cases (Stevens 1995).
S. pyogenes occurs in 80 different varieties, many of which produce
slightly altered versions of the SPE toxins. Cross-infection of different
strains of S. pyogenes by bacteriophages can lead to a phenomenon
similar to antigenic shift in viruses that leads to cyclic outbreaks of
highly virulent S. pyogenes. The emergence of TSLS in the
late 1980s may represent such an episode. In fact, the sudden rise
in incidence of soft-tissue necrosis caused by TSLS led to substantial
tabloid media coverage of a "flesh-eating bacteria epidemic" in the late
1980s and early 1990s (Stevens 1995).
Toxins involved in TSS and TSLS
The various toxins related to TSS and TSLS are structurally homologous
to varying degrees. The SEs are strongly homologous to each other,
as are the SPEs, and SEs and SPEs are homologous to each other to a lesser
extent (Bohach et al. 1990). TSST-1 possess the least structural
homology to the other toxins -- only 30%. TSST-1 is also the only
pyrogenic toxin that does not require a zinc cofactor (Schmitt et al.
1999). SE B, SE C, and SPE A share the most structural similarity
and are also relatively cross-reactive to the same antibodies. Interestingly,
the three toxins have the most antibody cross-reactivity at their amino
termini but share the most structural homology at their carboxy termini.
Several investigations have also produced conflicting data regarding the
identity of the terminus responsible for toxic activity (Bohach et al.
1990). Biochemical data for all the toxins are summarized in Table
1.
Table 1. Biochemical data for TSS and TSLS toxins.
Amino acid and MW data are for mature protein (after signal sequence cleavage),
MWs are predicted from amino acid sequence. All data are from Bohach
et al. (1990).
|
Name
|
Source
|
Number of amino acids
|
Predicted MW
|
Comments
|
|
TSST-1
|
Staphylococcus aureus
|
194
|
22,049
|
25% hydrophobic, but stable in aqueous and basic solution
|
|
SE B
|
Staphylococcus aureus
|
239
|
28,336
|
resistant to pH, temperature, and proteolytic degradation
|
|
SE C
|
Staphylococcus aureus
|
239
|
25,531
|
resistant to pH, temperature, and proteolytic degradation
|
|
SPE A
|
Streptococcus pyogenes
|
221
|
25,787
|
acidic
|
|
SPE B
|
Streptococcus pyogenes
|
?
|
~29,000
|
basic
|
|
SPE C
|
Streptococcus pyogenes
|
208
|
24,354
|
acidic
|
Superantigenicity
All of the TSS- and TSLS-related toxins are able to function as superantigens:
proteins that simultaneously bind nonspecifically to T cell receptors (TCRs)
and MHC class II molecules outside of the normal peptide-binding groove
(Fig. 1; Bohach et al. 1990). TSST-1, the best characterized
of the toxins, contains two structurally distinct binding domains, a b-grasp
motif that binds to the Vb CDR2 loop
and a b-barrel oligosaccharide/oligonucleotide
fold that binds to the MHC class II molecule (Schmitt et al. 1999),
possibly using hydrogen bonds (Kum et al. 2000). Each superantigen
exhibits relatively broad specificity for MHC class II molecules, but can
only bind to a few (1-5) Vb allotypes
(Schlievert 1993).
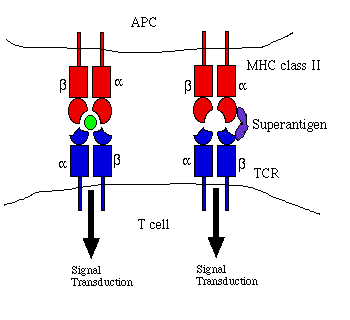
Figure 1. Comparison of antigen and superantigen
binding to TCR : MHC class II complex. Left side: normal antigen
presentation. Right side: superantigen stimulation in absence of
antigen recognition. Red = MHC, blue = TCR, green = antigen, purple
= superantigen. Adapted from Janeway et al. (1999) and Schlievert
(1993).
Superantigen binding of TCRs and MHC class IIs on antigen presenting
cells (APCs) activates the T lymphocyte. Because superantigens bind
nonspecifically, polyclonal populations of CD4 T cells are activated and
begin proliferation (Schlievert 1993). Such nonspecific T cell proliferation
may be induced by as little as 100 pg/ml of superantigen (Bohach et
al.1990).
The large numbers of effector CD4 T cells resulting from this nonspecific
proliferation begin stimulating monocytes to secrete several cytokines,
including tumor necrosis factor (TNF) a and
interleukin (IL) 1. The nonspecific, high volume superantigen stimulation
of T cells results in systemic secretion of these cytokines (instead of
the localized secretion that normally occurs during infection), which causes
much of the morbidity associated with TSS and TSLS (Janeway et al.
1999, Schlievert 1993).
Some data suggest alternate pathways for TSST-1 stimulation of T cell
proliferation. When APCs were incubated with TSST-1 for 24 h, then
washed extensively and introduced to T cells, the APCs were still able
to induce T cell proliferation. This result suggests some sort of
internalization and MHC-related presentation of the superantigen, rather
than the traditional extracellular superantigen mechanism. The results
of another investigation indicate that TSST-1 activates the IP3
pathway in a manner similar to that of lectin mitogens (Bohach et al.
1990). Additionally, TSST-1 may be able to stimulate monocyte secretion
of cytokines in the absence of T cell proliferation. When T cell
proliferation was inhibited with cyclosporin, TSST-1 still induced TNF-a
production in a lymphocyte/monocyte culture (Schlievert 1993).
TSS/TSLS Symptoms and Proximal
Causes
The primary symptoms of TSS and TSLS are listed below. Both syndromes
have exceptionally short incubation times, and most of these symptoms will
manifest within a matter of 8-12 hours after infection (Chesney 1989).
Effects are summarized in Fig. 2.
Fever. TNF-a and IL-1 secreted
by monocytes act on the hypothalamus to raise the bodily thermostat and
cause fever. TSST-1 may also cross the blood-brain barrier and interact
with the hypothalamus directly to induce fever (Bohach et al. 1990).
Susceptibility to Endotoxins. The pyrogenic toxins increase
the host's susceptibility to the endotoxins of other bacteria 105-106-fold.
In the presence of TSS and TSLS exotoxins, only a few picograms of endotoxin
are necessary to induce further shock. Such low levels of endotoxin
may be introduced by Gram-negative bacteria already present in the vagina
and intestine. Additionally, SPE A binds to endotoxins, forming a
complex that is lethal to immune cells (Schlievert 1993).
Hypotension. Severe systemic hypotension is caused by a
reduction in vasomotor tone, which allows blood to pool in the peripheral
tissues, and increased capillary leakage. Fluid leakage out of the
vasculature decreases blood volume and lowers blood pressure. Leakage
is though to be caused one of three mechanisms (or a combination thereof):
1) TNF-a and IL-1 promote vascular permeability,
allowing fluid to leak into tissue interstices. 2) TSST-1 alters
capillary permeability directly. 3) Endotoxins (with amplified
lethality due to action of pyrogenic toxins) cause capillary leakage directly
or indirectly (Schlievert 1993). Forni et al. (1995) suggest
that in TSLS, most hypotension is actually a result of reduced cardiac
output rather than changes in the systemic vasculature.
Diarrhea. Severe diarrhea appears to be mediated directly
by pyrogenic toxins (Chesney 1989).
Multiple Organ System Failure. Reduced tissue perfusion
caused by systemic hypotension causes problems for many organs, including
the brain, where cerebral edema leads to headaches, disorientation,
and confusion. In a clinical setting, reduced blood volume and dehydration
from diarrhea and vomiting require massive fluid replacement that eventually
damages the kidneys, causing renal failure, and in some cases respiratory
distress. Impaired circulation to the heart will also cause myocardial
failure and death (Chesney 1989).
Erythroderma (rash). Vascular dilation leads to redness
of the epithelia, especially the conjunctiva and sclera of the eye, which
may hemorrhage (Chesney 1989). Pyrogenic toxins are also able to
induce degranulation of mast cells and upregulate IgE production (Schlievert
1993); such effects would magnify type I hypersensitivity (Janeway et
al. 1999). TSS also produces long term dermal effects, including
desquamation (skin peeling) on the hands and feet after 10-21 days and
hair and nail loss after 4-6 weeks (Chesney 1989).
Humoral Immunosuppression. Despite the size and apparent
antigenicity of TSST-1, many patients (nearly 85%) suffer TSS repeatedy
without ever mounting a humoral immune response to TSST-1 (Schlievert 1993),
and TSS patients exhibit reduced IgG serum levels (Bohach et al.
1990). Superantigenic T cell activation by TSST-1 appears to stimulate
widespread production of interferon (IFN) g
by T lymphocytes. IFN-g has been shown
to inhibit Ig production and could explain the observed humoral immunosuppression:
B cells are not stimulated to produce Ig and never mount a primary immune
response (Schlievert 1993). This theory seems to be widely accepted,
though in mice IFN-g actually stimulates isotype
switching to IgG3 and IgG2a (Janeway et al. 1999).
Lack of Purulence. The widespread release of TNF-a
seems to inhibit the chemotactic capabilities of neutrophils and no localized
inflammation or purulence (pus formation) is seen in the area of initial
bacterial infection (Bohach et al. 1990).
Necrotizing Fasciitis (TSLS only). Streptococcus pyogenes
infection usually causes severe, gangrenous necrosis of subcutaneous fascia
and fat tissue, though the surrounding skin and muscle is usually unharmed
(Stevens 1995).
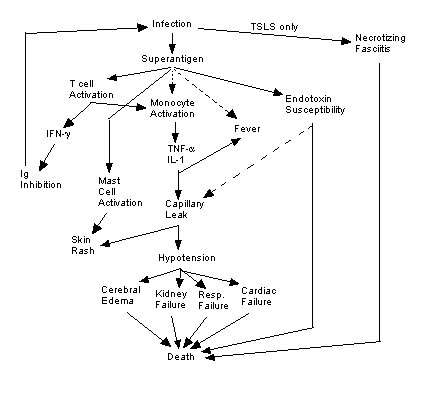
Figure 2. Summary of causes and effects of TSS
and TSLS. Solid lines indicate known effects, broken lines indicate
putative relationships. Image created by author.
Case Definitions of TSS
and TSLS
Because no lab tests are available to confirm diagnosis of TSS or TSLS,
clinicians must rely on the following standard case definitions (Chesney
1989).
Staphylococcal TSS (Deresiewicz 1999):
| Category |
Threshold |
| Fever |
* 38.9 °C |
| Rash |
Diffuse macular erythroderma ("sunburn") |
| Hypotension |
Systolic bp ? 90 mm Hg |
| Desquamation |
1-2 weeks after onset |
| Multisystem Dysfunction (at least 3 of the following) |
|
| Gastrointestinal |
Vomiting / diarrhea |
| Muscular |
Myalgieas or serum creatine phosphokinase ? 2 x normal level |
| Mucous membranes |
Vaginal, oropharyngeal, or conjunctival hyperemia |
| Renal |
Blood urea nitrogen or creatinine * 2 x normal; pyuria |
| Hepatic |
Total serum bilirubin or transaminase * 2 x normal |
| Hematologic |
Platelets ? 100,000 / liter |
| CNS |
disorientation or altered consciousness |
Streptococcal TSLS (Stevens 1995):
A. Isolation of group A Streptococcus
1. from a sterile site
2. from a nonsterile site
B. Clinical signs of severity
1. Hypotension
2. Clinical and laboratory abnormality (2
or more of following):
a. renal impairment
b. coagulopathy
c. liver abnormalities
d. acute respiratory
distress syndrome
e. necrotizing fasciitis
f. erythrematous rash
Definite case = A1 + B(1 + 2)
Probable case = A2 + B(1 + 2)
Suggested Treatments
Treatment for both TSS and TSLS involve the same basic steps:
1) Clean any obvious wounds and remove any foreign bodies (Chesney 1989).
2) Prescription of appropriate antibiotics to eliminate bacteria (Chesney
1989); Stevens (1985) recommends penicillin and clindomycin for S. pyogenes.
3) Monitor and manage all other symptoms, e.g. administer IV fluids
(Chesney 1989).
4) For severe cases, administer methylprednisone, a corticosteriod inhibitor
of TNF-a synthesis (Bohach et al. 1990).
5) For TSLS, debride necrotizing fascia in timely manner; perform fasciotomy
or amputation as necessary (Stevens 1995).
Experimental / Future Treatments
One possible treatment for severe TSS is IV delivery of pooled anti-TSST-1
IgG. IgG treatment will neutralize TSST-1 toxin, but it will also
inhibit the production of a native primary immune response and the formation
of immunological memory for TSST-1 (Chesney 1989) due to the antibody-mediated
suppression of naive lymphocyte activation (Janeway et al. 1999).
Therefore, IV Ig treatment is only recommended for severe or recurrent
cases of TSS (Chesney 1989).
As of 1989, no antitoxins were recommended for use in TSS patients (Chesney
1989), but in April 2000 several Isreali researchers reported the development
of an agonist peptide for TSST-1 that prevented TSS in 100% of mice when
administered before TSST-1 inoculation and rescued 50% of mice from TSS
when administered after onset of TSS (Siegel-Itzkovich 2000). Of
course, clinical trials will be needed to determine the effectiveness of
such agonists in treating human TSS.
Literature Cited
Bohach GA, Fast DJ, Nelson RD, Schlievert PM. 1990. Staphylococcal
and streptococcal pyrogenic toxins involved in toxic shock syndrome and
related illnesses. Critical Reviews in Microbiology 17(4): 251-272.
Chesney PJ. 1989. Clinical aspects and spectrum of illness
of toxic shock syndrome: Overview. Reviews of Infectious Diseases
11(supplement 1): S1-S7.
Deresiewicz RL. 1999. Toxic shock syndrome: a health professional's
guide. <http://www.toxicshock.com/healthprof.htm>
Accessed 2000 20 April.
Forni AL, Kaplan EL, Schlievert PM, Roberts RB. 1995. Clinical
and microbiological characteristics of severe group A streptococcus infections
and streptococcal toxic shock syndrome. Clinical Infectious Diseases
21: 333-340.
Janeway CA, Travers P, Walport M, Capra JD. 1999. Immunobiology:
the immune system in health and disease. 4th ed. New York:
Elsevier Science, Ltd/Garland Publishing.
Kum WWS, Laupland KB, Chow AW. 2000. Defining a novel domain
of staphylococcal toxic shock syndrome toxin-1 critical for major histocompatibility
complex class II binding, superantigenic activity, and lethality.
Canadian Journal of Microbiology 46(2): 171-179.
Schlievert PM. 1993. Role of superantigens in human disease.
Journal of Infectious Diseases 167: 997-1002.
Schmitt CK, Meysick KC, O'Brien AD. 1999. Bacterial toxins:
friends or foes? Emerging Infectious Diseases 5(2). <http://www.cdc.gov/ncidod/_vti_bin/shtml.dll/EID/vol5no2/schmitt.htm/map>
Accessed 2000 20 April.
Siegel-Itzkovich J. 2000. Scientists build a peptide to
stop toxic shock syndrome. British Medical Journal 320: 958.
<http://www.bmj.com/cgi/content/full/320/7240/958>
Accessed 2000 20 April.
Stevens DL. 1995. Streptococcal toxic-shock syndrome: Spectrum
of disease, pathogenesis, and new concepts in treatment. Emerging
Infectious Diseases 1(3) 69-78.
Return to Will White's homepage
Return
to Immunology Homepage

For questions or comments, please contact wiwhite@davidson.edu.
Copyright 2000, Davidson College Department of Biology,
Davidson, NC 28036.