This web page was created as an assignment for an undergraduate course at Davidson College.
Dermatomyositis
What is Dermatomyositis?
Dermatomyositis (DM) is an acquried muscle disease, also known as an inflammatory myopathy. While not particularly well-understood and extremely varied in its causes, symptoms, and outcomes, it is generally characterized by a rash that accompanies or precedes muscle weakness (NINDS, 2001). Although a man named Unverricht first described the disease in 1887 (Ramanan, 2002), it was not well-characterized until 1975 when a set of five criteria were established to aid in classification of the disease: progressive proximal symmetrical muscle weakness, elevated muscle enzymes, abnormality on an electromyogram (EMG), abnormal muscle biopsy, and cutaneous skin disease (Callen, 2002).
Who gets Dermatomyositis?
Dermatomyositis affects both children and adults, but tends to affect females more often than it affects males (2:1) (NINDS, 2001 and Merck, 2003). In the United States, the incidence of DM is estimated at 5.5 cases per million people (Callen, 2002). If there are approximately 290 million people in the United States, then about 1595 people have this disease, making it extremely rare. The disease usually affects adults after the second decade of life (Brasington, 2003). Juvenile DM is even rarer, with average onset between 8-9 years of age (Juvenile, 1998). However some investigators found that there were two peaks of onset for females, occurring at age 6 and at age 11, whereas males usually acquired the disease prior to the age of 10 (Symmons, 1995). Again, among European ancestries, juvenile DM is about twice as common in girls than in boys. The data from Japanese and Saudi Arabian studies show the opposite effect: a 1:2 female to male ratio (Ramanan, 2002). These differences suggest that genetics must play an important role in the susceptibility of disease.
What are the symptoms of Dermatomyositis?
Skin and muscle involvement are the most common symptoms of DM, but it may affect other organ systems as well. The skin rash is generally patchy, violet discolorations on the face, neck, shoulders, upper chest, elbows, knees, knuckles, and back. The lilac, erythematous heliotrope rash generally surrounds the eyes and is highly characteristic of DM because it is rarely observed in other disorders (Callen, 2002). In other words, patients tend to look like they have raccoon eyes. To see heliotrope rash, go here. Another cutaneous manifestation includes Gottron's lesions, which are erythematous, scaly lesions that cover certain joints like the knees and elbows (Ranaman, 2002). Many other images pertaining to the skin rash in dermatomyositis can be seen at this site.
Muscle weakness is the most common symptom, with muscles closest to the trunk of the body (proximal) being affected most often. Patients will have trouble rising from a sitting position, climbing stairs, lifting objects, or reaching overhead. Muscles will ache and be tender to the touch, and in some cases, swallowing will become difficult (dysphagia). Other systemic muscle symptoms include joint swelling, the Raynaud phenomenon and cardiopulmonary abnormalities (Callen, 2002). Most patients will experience a reduction in ventilatory capacity due to muscle weakness in the lungs. Patients also develop Inflammation of the joints in the form of arthritis at some point in the course of the disease. Luckily, this arthritis is usually non-deforming and non-destructive (Ramanan, 2002). Finally, patients will feel fatigue and discomfort and may lose weight and/or have a fever (NINDS, 2001).
Aside from the distinct heliotrope rash and the marked muscle degeneration and weakness, DM can be diagnosed by looking at abnormal electromyogram (EMG) readings and the result of muscle biopsies. The EMG is an electrical way to test nerves and muscles. EMG findings should demonstrate irritability upon insertion of the needle and short-duration, low-amplitude, polyphasic potentials. The muscle biopsy is most helpful in determining the type of DM by showing specific antibodies and other enzymes in the muscle (Chitnis, 2003).
What causes Dermatomyositis?
Like many autoimmune diseases, the cause of this dermatomyositis is unknown. Lab studies have found that muscle enzyme levels are often abnormal in both juvenile and adult patients. Studies have shown that the muscle enzyme creatine kinase (CK) is the most commonly found elevated enzyme. CK is not normally found in the bloodstream; thus its increased presence indicates damage to the muscle or to the brain (Corbett, 1987). Obviously then, these elevated enzyme levels are not the cause of the disease, but rather the result of muscle damage due to some other cause.
It is very likely that a genetic predisposition to dermatomyositis exists and that it may be linked to certain human leukocyte antigen (HLA) types, but no evidence as yet has been found to support that nor have any particular genotypes been elucidated. Abnormal T-cell activity seems to be involved in the pathogenesis of both the skin and the muscle disease. However, the presence of T cell muscle infiltrates in DM patients does not lead to a detectable repertoire in the blood (Benveniste, 2001). Other immunological abnormalities are quite common in patients however. In particular, autoantibodies to nuclear antigens (ANAs) and cytoplasmic antigens maybe be present. Their presence do help to define particular subtypes of DM or polymyositis (which affects just muscles), the role of these autoantibodies in pathogenesis is unclear (Callen, 2002). One study suggested that DM is completely antibody-mediated. Immunofluorescence studies have shown that there is immune complex deposition in the endothelium. PM, on the other hand, is the truly T-cell mediated disease. Investigators have shown that it is characterized by an inflammatory infiltrate containing mostly CD8+ T-cells (Chitnis, 2003).
The autoantibodies found in DM appear to be nonspecific for connective tissue disease. In fact, viruses such as coxsackievirus B are sometimes implicated in the pathogenesis of the disease. Patients can also have anti-Jo-1 antibodies to the histidyl transfer RNA synthetase (Chitnis, 2003). Anti-Mi-2 is another antibody present in individuals with DM, and it is known to target a helicase involved in regulating transcription (Brasington,2003). Why then do these antibodies only seem to attack this helicase in muscle cells and skin cells instead of in all cells of the body? Again, little is known about the actual method of destruction. These high levels of autoantibodies are against intracellular antigens and are found in the absence of an antibody-induced inflammation. How do these antibodies enter living cells? Some think that they are actually simply markers of a particular pattern of tissue injury and that they do not contribute to the immunopathology of the disease itself (Janeway, 2001). In that case, the antibodies are being made in response to this tissue injury, this necrosis, which would explain their elevated levels. However, muscle biopsy specimens from patients with DM show that there is an infiltration of the B-cells targeting the microvasculature, and tissue damage itself seems to be the result of this immunologic activation.
Some autoimmune reactions that damage tissues are similar to type II hypersensitivity reactions. IgG or IgM responses to autoantigens that are located on the cell surface of muscle and skin (in the case of dermatomyositis) cause the injury. The antibody bound to the cell will induce the cell's destruction by macrophages or natural killer (NK) cells (Janeway, 2001). It is important to note that nucleated cells are generally resistant to lysis by the complement mechanism, but the membrane-attack complex can still form on these cells. These antibodies can bind to C1q, which is one of the first proteins involved in the classical pathway of complement activation. This binding of C1q thus activates the entire complement cascade, ending in the formation of the membrane attack complex, which forms a pore in the cell membrane, disrupting the cell's integrity and eventually killing it (Janeway, 2001).
In juvenile DM, deposits of IgM, IgG, and the third component of complement have been found in the walls of skeletal muscle (Merck, 2003). These complexes appear in patients who have a deficiency in the complement system (generally a genetic defect) and cannot therefore effectively clear immune complexes, causing tissue damage (Janeway, 2001). For example, some patients with DM have C9 deficiency (Ichikawa, 2001). C9 is the molecule that polymerizes to form a pore in the membrane; if this pore is not made, then the proper immune response is never initiated. There is then a buildup of immune complexes, which can cause tissue damage.
Unfortunately, much of this antibody activation and genetic susceptibility only comes into play once the immune system has been activated. The true causes of the disease are unknown and are probably associated with infection.
How is Dermatomyositis treated?
Before the use of corticosteroids, about one third of patients with DM died of their illness and about one third of them developed permanent severe physical limitations. The last third seemed to recover spontaneously. In the 1970s and 1980s, there was about a 10% decrease in mortality, but it was not until corticosteroids were safe to use on a regular basis that DM could be cured more immediately (Taieb, 1985).
The steroid of choice is prednisone and doses of up to 100 mg/day can be used initially to curb the response. The duration of the medication depends completely on its ability to control the disease and how quickly it is able to do that (Brasington, 2003).
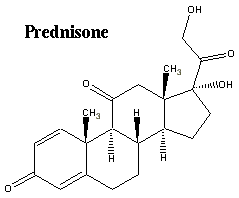 Structure of prednisone. Image from http://www.sciencesoft.net/prednisone.html.
Structure of prednisone. Image from http://www.sciencesoft.net/prednisone.html.
Prednisone is a lipid-soluble molecule that can cross the phospholipid bilayers of cell membranes. Therefore, it does not discriminate which cells it enters. While this is beneficial because it can enter those cells that are eliciting the immune response. It enters the cell and binds to the steroid receptor in the cytosol. Once it binds, HSP90 (a heat shock protein bound to the DNA binding domain of the steroid receptor) is released. The DNA binding domain is exposed and the steroid:receptor complex enters the nucleus and binds to promoter regions of certain genes, inducing their transcription. Usually they can decrease inflammtion caused by cytokines and decrease the number of prostaglandins and leukotrienes. Unfortunately, prednisone acts on many genes not associated with the immune system and many side effects are manifested, including weight gain, fluid retention, mood swings, bone mineral loss, and thinning of the skin (Janeway, 2001).
Methotrexate and azathioprine are also used in replace of steroids, but they do not have quite the same effect (Brasington, 2003).
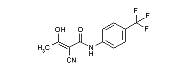
Structure of azathioprine. Image from http://www.prous.com/journals/dot/sample/html/DT360395/DT360395.html#azathioprine.
Azathioprine is able to interfere with DNA synthesis by blocking the pathway of purine synthesis. Without purines, DNA cannot be synthesized or transcribed. Therefore, this drug acts best on dividing cells (Janeway, 2001). In many instances, physicians may treat patients with a combination of azathioprine and prednisone in order to maximize response while minimizing side effects.
Mortality rates now vary from 15%-35% and death is usually due to cardiac or pulmonary failure (Chitnis, 2003).
Images of Dermatomyositis
Due to copyright and permission difficulties, no images were able to be obtained for the creation of this webpage. However, many images can be found at the below websites:
Images from the University of Iowa