This web page was produced as an assignment for an undergraduate course at Davidson College.
Metagenomics
What is Metagenomics?
Metagenomics is a growing field in genomics that does not require the cultivation of microbes and analyzes the genomes of samples that are taken straight from the environment. The DNA from these samples is extracted and sequenced and the data is analyzed using computational tools (Banfield & Sharon, 2013). The purpose of metagenomics is to collect information regarding the physiology and genetics of uncultured organisms
Advantages to Metagenomics?
Methods
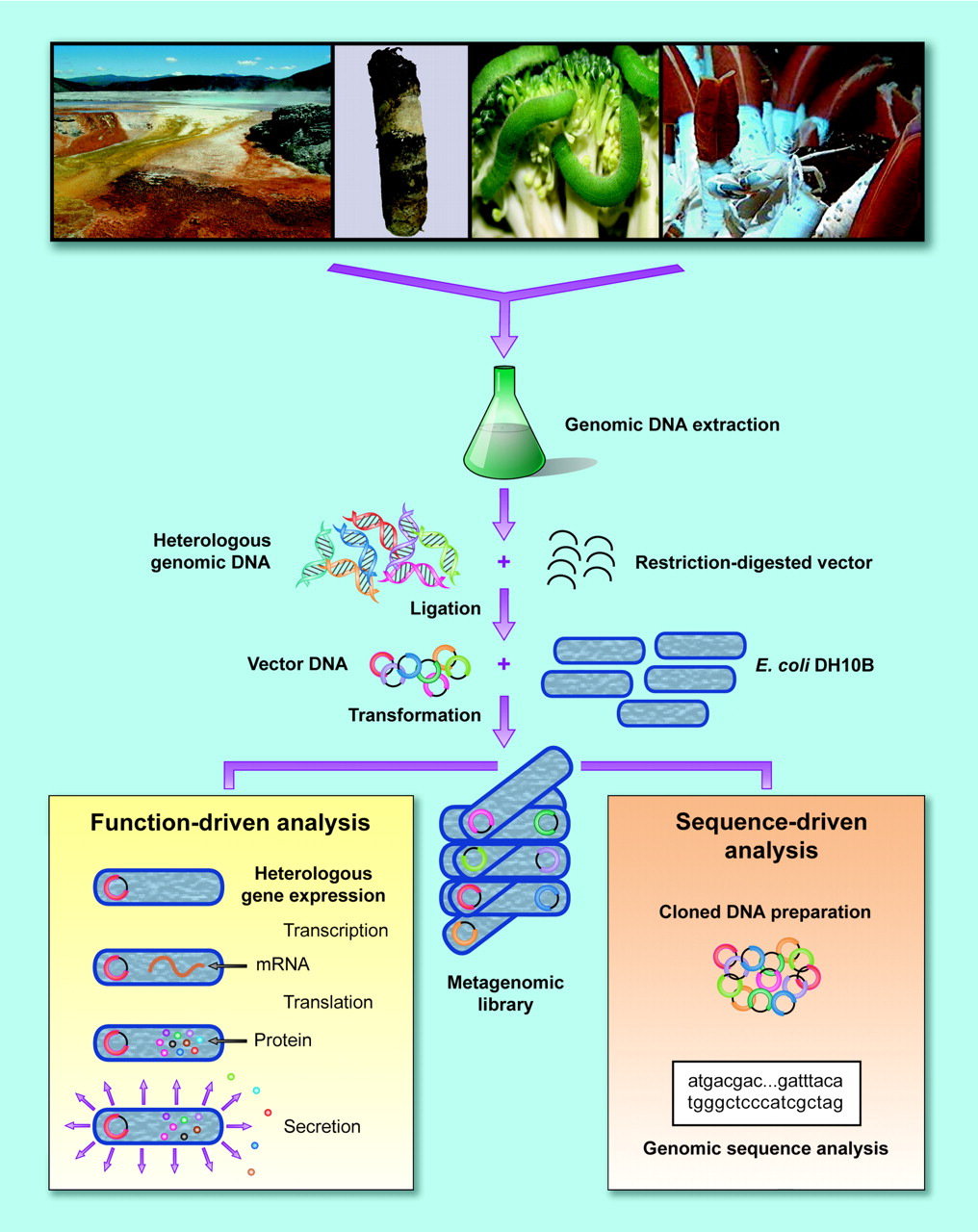
The metagenomic analysis of microbial genomes involves several steps with varying approaches to analzying a genome. First, DNA is isolated from the environment, then cloned into a vector. Then the clones are transformed into host bacteria and the subsequent transformations are screened (Handelsman, 2004).
Screening
Clones can be identified, or screened, using traits that are unique to that particular organism or sequence. The following are some items that are screened for:
Figure 1: A visual representation of the various steps and different approaches in metagenomics. (Handelsman, 2004. Permission Pending)
There are then two main approaches to the analysis of a genome: sequenced-based analysis and functional metagenomics.
Sequenced-Based Analysis
Sequenced-based analysis uses the sequences of genomic fragements in order to identify the clone of orign. It is a very powerful tool used to determine linkage of traits, the organization of an organism's genome, horizontal gene transfer, and distribution and redundancy of functions in a community. From this analysis method scientists can find relationships between phylogeny and function. However, sequenced-based analysis has its limitations as well. Not every gene of interest or DNA fragment has a phylogenetic marker, since there are only a few available markers that allow for the accurate placement of genomic fragments in the "Tree of Life" (Handelsman, 2004). When such genomic fragments are sequenced, the organisms of origin cannot be determined using sequenced-based analysis.
Complete Sequencing - Here the genome is sequenced and the phylogenetic markers are used to identify the taxonomic group of the organism whose fragments were analyzed. This method is most useful when studying genomic fragments within a taxon. Although sophisticated environments and taxa may make it impossible to reconstruct the genome of an organism using complete sequencing, the genomic data sheds light on physiological and ecological features of organisms in the sample.
Random Sequencing - While most effective when done on a large scale, this method consists of sequencing the genome and identifying a gene of interest. Afterwards, the phylogeny of the gene is determined by searching for phylogenetic markers in the flanking DNA of the gene.
Functional Metagenomics
In this approach to metagenomic analysis, the function of a gene's products is used to identify the clone of origin. In order to identify an organism by its gene's protein, the protein must first be made. This means that functional analysis necessitates the transcription and translation of a particular gene and then ensuring that the product is secreted if it is usually found on the outside of the cell. This approach does not depend on the sequence of a fragment to identify a gene, making the method fundamental in identifying new classes of genes for functions that can be either known or unknown. However, a major downfall to functional genomics is related to the bacterium chosen for cloning. No matter the bacterium chosen, the majority of genes from the organism of interest will not be expressed. Without the expression of these genes, the functional analysis method crumbles since there are no gene products to analyze (Handelsman, 2004).
Future & Problems
The future and growth of metagenomics is heavily dependent on developments in bioinformatics. One of the areas of growth neccesary is new algorithms which account for the presence of multiple genomes that differ in coverage and degree of relatedness (CBCB, 2014). The traditional methods used in genomics are insufficient because scientists focus on specific features of all bacterial genomes, unlike the data in metagenomics which consists of multiple genes in different genomes. The nature of the data demands that new methods in bioinformatics develop in order to identify organisms by their genomes.
In addition, metagenomics will grow significantly as the database for phylogenetic markers grows. As the amount and types of anchors increase, the utility of sequenced-based analysis will also increase since it relies on phylogenetic markers to identify the organism of origin. With a more effective analysis approach, fragments that previously remained unidentified can be matched to their respective organism, leading to an eventual completion of a reconstructed genome for that organism. The significance of a reconstructed genome is that even more phylogenetic markers can be associated with a gene, further increasing the utility of random sequencing since a greater number of genes will now have phylogenetic markers in their flanking DNA (Handelsman, 2004).
Lastly, a significant portion of the future of metagenomics is the use of functional anchors. These anchors are functions that can be quickly surveyed in all of the clones in a library. Once there is a significant number of clones that have common functional markers, the clones can be analyzed for phylogenetic markers in the flanking DNA. This would allow for scientists to study the diversity of genomes that share a function while the respective genes can all be expressed in that host. This new approach to metagenomic analysis can only progress with developments in technology used in functional expression and screening (Handelsman, 2004).
References
Banfield, J., Sharon I. 2013. Genomes from Metagenomics. Science 342:1057-1058.
Email Questions or Comments to gacambronero@davidson.edu
© Copyright 2014 Department of Biology, Davidson College, Davidson, NC 28035