This web page was produced as an assignment for an undergraduate course at Davidson College.
JAMES HARDEN's
Genomics Web Page
Single-Cell RNA-Seq Reveals Dynamic, Random Monoallelic Gene Expression in Mammalian Cells.
The purpose of this article is to give insight and counter-evidence to the misconception that transcription of autosomal genes occurs from both parental alleles. Generally, researchers study the allelic expression of diploid cell populations, leaving out many necessary studies on allelic expression pattersn within a single cell. Therefore, these researchrs analyze allelic expression across individual cells of mouse embryos (CASTxC57). They found independent occurences of monoallelic expression of autosomal genes, and found great variation from cell to cell within a given embryo. Additionally, these same patterns were found in mature cells.
I personally feel that this paper was hard to follow because there was much supplemental information that was left out. Though their overall points were clear, the data was quite complicated and lacked adequate explanations of their significance. In addition to having ill-explained results, I felt that many of the methodologies were also skimmed over, which includes the fact that there is no methods section for clarity. I suspect that these authors had too much information and was limited by journal stipulations on word count and length. However, I found the material to be really interesting. I was intrigued by the normative beliefs regarding transcription of autosomal genes that these authors challenged in this paper. I found the overall points to be insightful and thought-provoking, including the implications that these findings have on genetic disorders and disease penetrance.
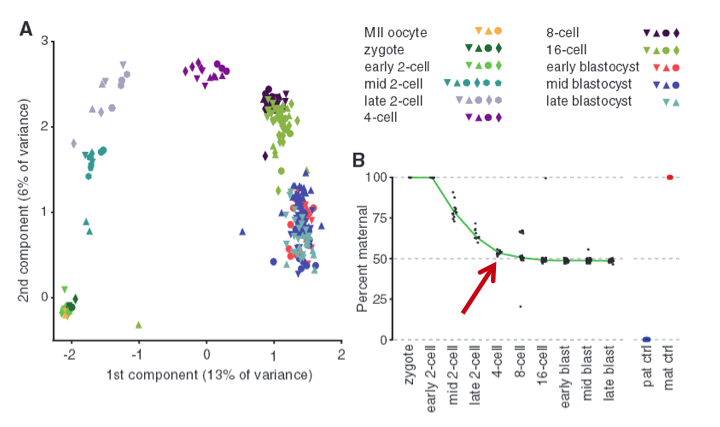
Figure 1 - Single-cell transcriptomes reconstruct preimplantation development.
To examine allele-specific gene expression at a single-cell resolution, they isolated 269 individual cells from F1 embryos of a CAST/EiJ x C57BL/6J cross (C57 is paternal and CAST is maternal). These cells ranged from oocyte to blastocysts stages. From these cells, they used Smart-Seq to make transcriptome profiles which they used to perform a principal component analysis (PCA) as shown in Fig. 1A. The important idea to take from this graph is the fact the data set is normally distributed, which lets us know that the two “components”, or the maternal/paternal alleles, are independently expressed (according to wikipedia definition of PCA).
To investigate further the allele-specific gene expression in a single cell, the researchers demonstrated that maternal genotype dominates in the transcriptome in the beginning stages and then reaches parity with paternal transcripts at the four-cell stage as shown in Fig. 1B (marked by red arrow). The data shows a pretty clean downward trending curve that flattens at 50 percent, with few outliers. The blue and orange dots are the two controls. Overall, we see how transcriptome analysis via RNA-Seq can be used to study the mRNA profile of embryos during development stages, and we can accurately determine which alleles genes are expressed during a given period of time.
Figure 2 - De novo paternal X-chromosome inactivation.
Next, the researchers focused solely on paternal X (Xp) chromosomes from males and females. They highlighted similarities in paternal autosomes and paternal X-chromosomes in females as shown in Fig. 2A. Here, Xp autosomes of males and females both reach a paternal/maternal ratio of 1, which is consistent with the findings in Fig. 1B. However, at 4-cell stage there is initial activation of Xp X-chromosomes, but then it declines, resulting in higher expression of maternal X chromosome (Xm) relative to paternal X chromosome (Xp). Additionally, the bottom portion of Fig. 2A shows the allele expression of Xist and demonstrates how Xp in females express more Xist than Xm in males or females (RPKM refers to allele-resolution gene expression). This is important because X chromosome inactivation initiates from the X-inactivation center (Xic) where Xist is transcribed; therefore, Xist expression correlates with X chromosome inactivation. Each point on the graph represents 3 to 28 cells.
Even though Figure 2A demonstrates higher expression of Xist for inactivated Xp in females, Figure 2B shows that Xp silencing is not simply a function of distance from Xic gene (indicated by vertical dashed line). It appears that the fraction maternal expression increases from 4-cell to 16-cell to early blastocyst (green, blue, and red respectively), though there is not any indication of significance or sample size. Regardless, the authors emphasized that Xp is silenced as development progresses. These data demonstrate the promising applications in inferring biological signals from allelic information in single-cell RNA sequencing. We can have confidence that RNA-Seq is an effective means in quantifying or characterizing mRNA profiles.
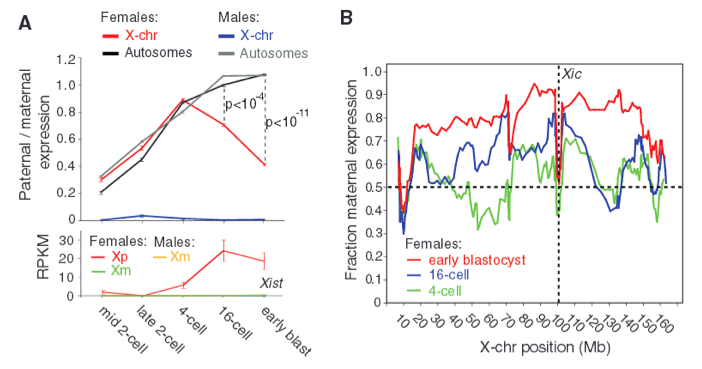
Figure 3 - Quantitative characterization of allelic transcription patterns.
The researchers were able to distinguish maternal and paternal monoallelic and biallelic expression using SNP-containing reads. However, single-cell transcriptome methods are subject to loss of RNA species; therefore, the researchers needed to determine how much random sampling effects inflate observed monoallelic calls. By lysing individual cells, splitting into two equal volume fractions, and independently processing them into sequencing libraries, the researchers were able to use split pairs to model the stochastic losses in RNA. I am not sure exactly how split pairs are used (making analysis of Fig. 3A-C difficult), but the authors emphasize the importance of using split cells to accurately analyze allelic expression in single cells. As such, the authors used the data in Fig. 3A to say that 60% of polyadenylated RNA is lost in the Smart-Seq process. As allele-resolution gene expression (RPKM) increases, the amount of allele loss decreases. Fig. 3B shows the inferred fraction of genes with monoallelic expression for genes binned by expression level. And Fig. C is the percent of genes with monoallelic expression for raw observation and the individual split cell experiments. Fig. 3C is not referenced in the article, and none of these 3 figures are explained in detail.
Researchers calculated that up to 66% of the observed monoallelic expression could be due to technical losses of RNA; therefore, they focused on transcripts expressing sufficient abundances which are less affected by random sampling. They were able to show the mean percent of genes with monoallelic expression across cells at each developmental stage as depicted in Fig. 3D. The maternal monoallelic expression (red) decreases as development progresses, as expected (Fig. 1B), and monoallelic expression is reflected in about 12-24% of the mRNAs from late 2-cell to late blastocyst stage (and this is apparently more monoallelic expression than would be expected). However, when researchers pooled for embryos, virtually all monoallelic expression is lost as shown in Fig. 3E. This highlights the fact that variation in monoallelic expression is found in the individual cell instead of a population of cells. This means that random monoallelic expression is cell-specific.
Furthermore, the researchers found that transcription of the two alleles occur independently, giving insight to the finding that, at any given point in time, every cell has random independent bursts of transcription. This is shown in Fig. 3F as the mean fraction of cells in 8-cell stage with biallelic expression for genes binned by their fraction of silent cells. Since this data suggest that monoallelic expression is an independent event, we would expect biallelic expression to have twice the mRNA as monoallelic expression, and this is in fact the case as shown in Fig. 3G. Fig. 3F and 3G together show that expression patterns are based on independent allelic transcription.
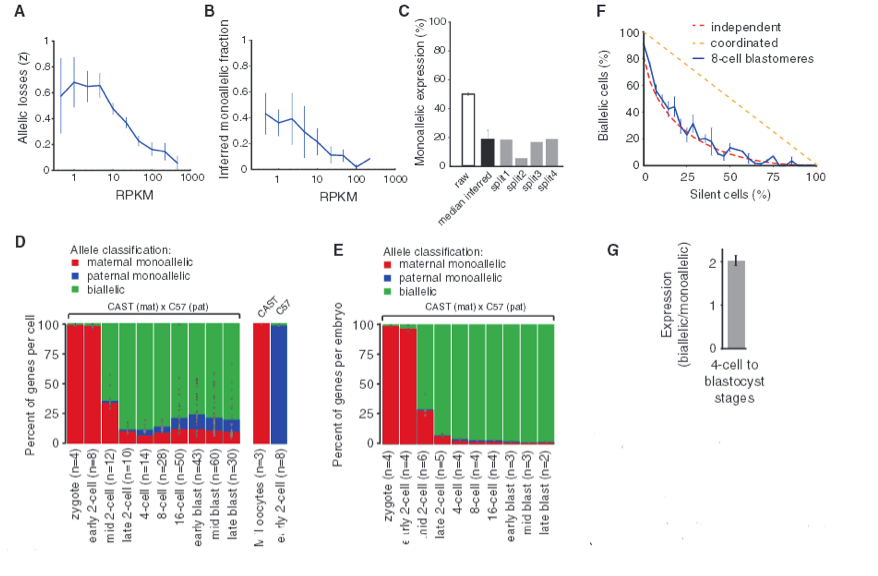
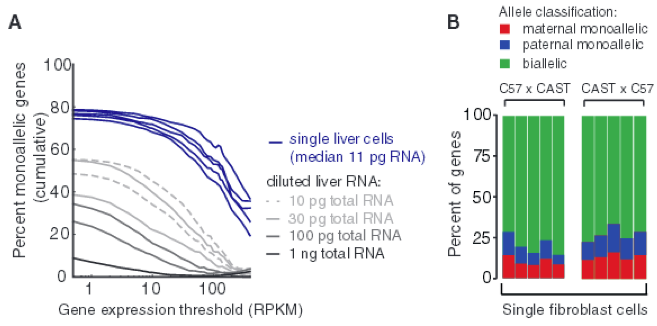
Figure 4 - Monoallelic expression in different mature cell types.
Lastly, it is useful to know if these observed expression patterns also exist in mature cells as well, so the investigators examined the single-cell transcriptomes of liver cells in C57 x CAST F1 crosses. As shown in Fig. 4A shows measurable percentage of monoallelic expression levels in mature cells when compared to the serial RNA dilution control. In addition, researchers obtained the percentage of genes with monoallelic expression in individual adult fibroblasts for genes that have at least 20 allele-resolution gene expression per stage, increasing the fraction of true monoallelic calls (Fig. 4B). Here, there is about 24% monoallelic expression per cell, on average. These two data sets together demonstrate that random monoallelic expression occurs in both embryonic and mature cells.
Main Points
The take home message for this article is that there are stochastic patterns of monoallelic expression that differ from allelic exclusion or regulation. Furthermore, though there is previous evidence/models of transcriptional bursting, this data suggest that this bursting happens independently for both alleles in each cell. Additionally, these findings could have fundamental implications in disease penetrance and severity. Overall, these results speak to the power of RNA-Seq and the capacity it has for producing mRNA profiles and characterizing transcriptomes on a cell to cell basis.
Genomics Page
Biology Home Page
© Copyright 2014 Department of Biology, Davidson College, Davidson, NC 28035