This web page was produced as an assignment for an undergraduate course at Davidson College.
JAMES HARDEN's
Genomics Web Page
What is Whole Transcriptome Shotgun Sequencing?
Whole Transcriptome Shotgun Sequencing is another name for RNA-sequencing and is part of the next-generation sequencing technology. Transcriptome sequencing includes RNA profiling, mRNA transcript expression analysis, and the sequencing and analysis of full-length mRNA transcripts (Ozsolak and Milos, 2011).
Why study the transcriptome instead of the genome?
DNA sequencing is the process of determining the nucleotide order of a given DNA fragment and RNA is much less stable inside the cell. Additionally RNA is generated by transcription from DNA, so it would seem that DNA and RNA provide the same information and studying the genome alone would suffice. However, because introns get excised, the RNA molecules do not necessarily parallel with the DNA template; therefore, we can gather information about gene expression from transcripts.
There are post-transcriptional modifications that occur such as 5’ capping, 3’ polyadenylation, and RNA splicing. These changes make the study of the transcriptome quite useful. It can be used to:
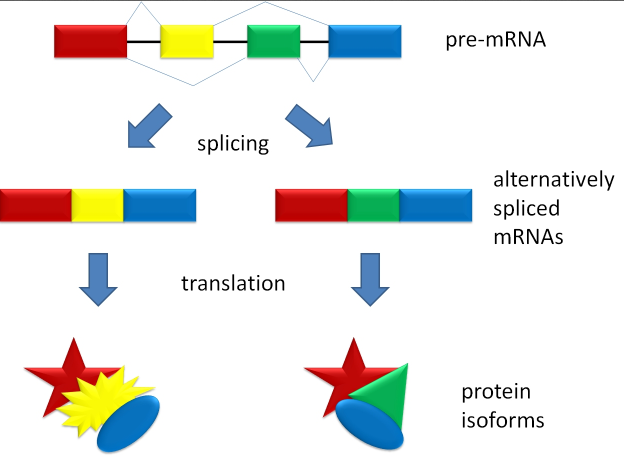
1) identify alternative gene spliced transcripts. A single gene can code for multiple proteins with alternative splicing. An analysis of the transcriptome can uncover transcripts produced via alternative splicing that would otherwise be unnoticed in the genome (Maher et al., 2009).
Courtesy of Wiki Commons
2) locate fusion genes or fusion transcripts. Gene fusion can result from two previously separate genes via translocation, deletion, or inversion, or it can occur post-transcriptionally as a result of trans-splicing (Maher et al., 2009; Nacu et al., 2011). Fusion genes and fusion transcripts are found in many types of cancers (Mitelman et al. 2007)
3) locate mutations and changes in gene expression. SNPs are single-nucleotide polymorphisms in the DNA sequence. SNPs that lead to changes in gene expression can be observed through transcriptome analysis (Maher et al., 2009).
4) examine different populations and types of RNA. With today’s technology, one can perform a size selection of target RNA molecules (Ingolia et al., 2012) and then perform Direct RNA Sequencing (developed by Helicos) (Raz et al., 2011).
5) determine exon/intron boundaries. Because introns are excised in transcripts, transcriptome sequencing makes it easier to determine where exons begin and end on the DNA strand. Additionally, one can confirm or contest previously annotated gene boundaries (Qian et al., 2013).
Method of RNA-sequencing.
Due to the nature of RNA, virtually all RNA sequencing methods parallel to each other, showing commonalties in the steps illustrated below.
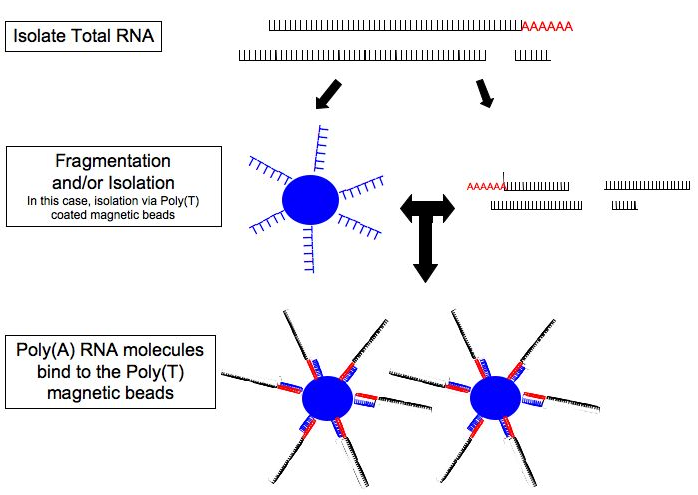
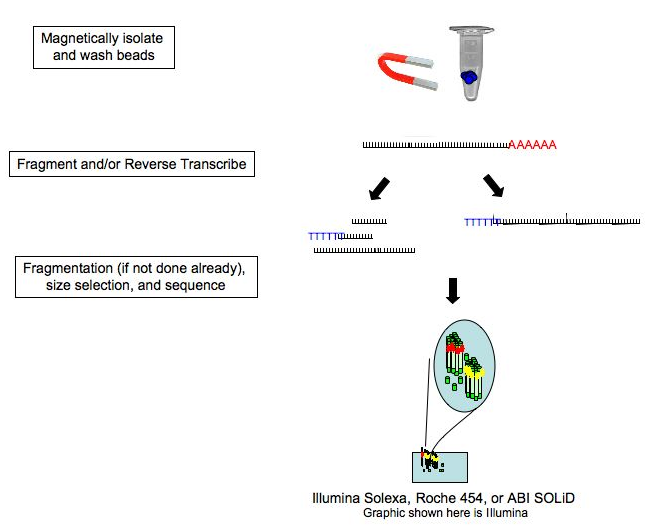
Courtesy of Wiki Commons
Comparison of methods in Transcriptomics
Prior to Next-Generation RNA sequencing, transcriptomics and gene expression studies were previously done with expression microarrays. Microarrays are based on probe hybridization, containing thousands of DNA sequences that probe for a match in the target sequence. Hybridization creates a profile of all transcripts being expressed. Transcriptome analysis was later performed by Serial Analysis of Gene Expression (SAGE), which tags cDNA sequences from mRNA transcripts and clones them into bacterial vectors for sequencing. A comparison of the methods is shown below.
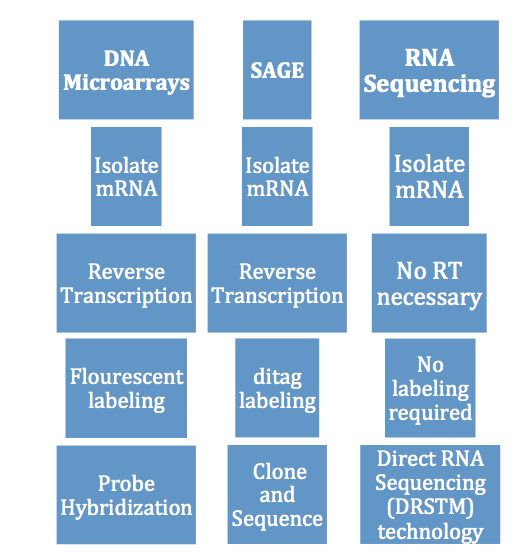
Produced by James Harden
To sequence RNA, the usual method is first to reverse transcribe the sample to generate cDNA fragments. However, this step is not needed in RNA sequencing and this can be beneficial because it eliminates insufficiencies with cDNA conversion such as incomplete conversion of 5’ and 3’ ends (Ozolak and Milos, 2011). In this respect, RNA sequencing is more advantageous than expression microarrays and SAGE.
RNA sequencing holds another advantage over microarrays specifically because it does not rely on cDNA libraries for probe hybridization. Microarrays are good for known common alleles, whereas RNA sequencing can uncover uncommon alleles. Additionally, microarrays are only as good as the SNP databases they’re designed from, so they have limited applications for rare mutations that go undetected. RNA sequencing has significant applications for cancer-causing mutations that are rare and undocumented (Ozsolak and Milos, 2011).
References
Assignment #2:You get to choose (due Feb. 28, 2 pm)
Assignment #3: You get to choose (due Apr. 25, 2 pm)
Genomics Page
Biology Home Page
© Copyright 2014 Department of Biology, Davidson College, Davidson, NC 28035