This web page was produced as an assignment for an undergraduate course at Davidson College.
The effects of Neandertal alleles in anatomically modern humans

Image courtesy of All you need is Biology.
Summary:
The focus of this paper was using electronic health records (EHR) to analyze the Neandertal variant contribution to approximately 28,000 derived phenotypes in adults of European ancestry using data from eMERGE.
A main complication of these types of analyses (on Neandertal DNA) is that confidently identifying Neandertal-derived DNA requires tests to determine trait association at specific sites between individuals with and without Neandertal ancestry. This problem was addressed using EHR with sufficient genomic data to determine phenotypes in 28,416 European adults and comparing these data to Neadertal alleles. The Neandertal alleles were determined based a comparison of recent genome-wide mapping of Neandertal haplotypes against individuals from the 1000 Genomes Project. This allowed the group to define 135,000 Neandertal SNPs for use.
To determine the impact of Neandertal variants on anatomically modern humans (AMH) phenotypes the group used genome-wide complex trait analysis (GCTA). The group used this method to quantify the overall influence of Neandertal SNPs together on traits in AMHs. These traits will be listed later in figure summary, but most notably actinic keratosis and depression. The group also compared phenotypic variance using a genetic relationship matrix (GRM) for the Neandertal SNPs against the non-Neandertal SNPs, which eliminated some of the less significant associations that had been identified. Then, using best linear unbiased predictions (BLUPs), the group found gene enrichment for regions associated with actinic keratosis (keratinocyte differentiation and immune functions) and depression (neurological diseases, cell migration, and circadian clock). The significant Neandertal allele enrichment near genes associated with long-term depression reinforces the notion that human-Neandertal DNA and methylation differences influence neurological and psychiatric disorders. The significant Neandertal allele enrichment near alleles associated with keratin filament formation and keratinocytes further reflect the influence of Neadertal DNA on AMH phenotypes.
To identify specific loci associated with a certain AMH phenotypes, rather than a genome wide association, the group performed a phenome-wide association study (PheWAS). Two meta-analyses with a locus-wise Bonferroni corrected significance threshold yielded 4 Neandertal SNP-Phenotype associations to the following: hypercoaguable state, protein-calorie malnutrition, urinary system symptoms, and tobacco use disorder. The group was able to identify and thoroughly explore specific loci responsible for risk associated with these phenotypes and then make informed speculations about AMH evolutionary history and the retention of these phenotypes.
The group's approach helped establish a new method for better understanding our evolutionary past, in this instance the genetic admixture between Neandertals and AMHs. This approach allowed the group to formulate many ideas and further our understanding of the influence of Neandertal alleles and AMH traits using EHR phenotypic data and linked genomic data. As genomic data becomes increasingly available linked to EHRs with phenotypes, this combination of EHR data paired with DNA sequencing data will become more and more useful, and this study lays a foundation for future research and methods.
Opinions:
I believe this group has begun to tap the potential of EHR data in a useful way. The group is restricted by the amount of genomic data available, but a sample size of ~28,000 seems relatively large and should provide robust representation.
The group continuously used different cohorts based on the phases of sample inclusion in the eMERGE Network Phases, which is I believe was a good methodological decision, since the two cohorts are based on two different data releases. Their results are subject to bias based on the populations in the 1000 genomes project data base as well as the Atlai Neandertal genome, but this would be true for any sort of meta-analysis of a data base, and hopefully the sample size provides robustness to their population.
When determining the significant associations in the data of the two cohorts, the group chose a P value of 0.1 to indicate a significant difference. Typically, a P value of .05 is used to indicate significance, and the reason for not using 0.05 is not explicitly explained. If the group had used a P value of 0.05, the P value for actinic keratosis in the E1 cohort would not be considered significant, and a great deal of time is spent exploring the significance of this particular association in detail. It seems reasonable that the group chose to use a P value of 0.1 rather than 0.05 to tell a more compelling story. I do believe the association findings are interesting.
Based on the group's findings, they suggest that some 1.15% - 1.06% of the risk for depression may be explained based on these Neandertal variants, and make similar claims for other disease phenotypes. While the group goes to great lengths to establish significance, clinical applications of this knowledge do not seem immediately useful. It is never explained exactly what is meant by “risk explained”, whether that is 1.15% - 1.06% of the entire population is depressed due solely to this variant, or if it is some small contributor in much of the population. This may be explained somewhere in the else, but I could not find it in the paper. Although current clinical applications may not present themselves, I believe the group achieved its goals of determining the influence of Neadertal alleles in AMH phenotypes. These significant findings may provide us with some insights into AMH evolutionary past and the group speculates more on some variants (those involved with depression) than others.
I think the true value of this paper is the approach and application of EHRs to make claims about human’s evolutionary history. These EHRs are becoming more and more prevalent and as genomic data becomes increasingly available, these types of studies have a huge potential for impact due to the incredible amount of information that can be gained. The group was able to explain a portion of the risk associated with Neandertal alleles (GTCA) and able identify specific loci with significant Neandertal SNP-phenotype associations (PheWAS). This approach will become even more powerful with each generation and we can establish electronic EHR lineages that will be able to inform us more and more.
Figure Summary
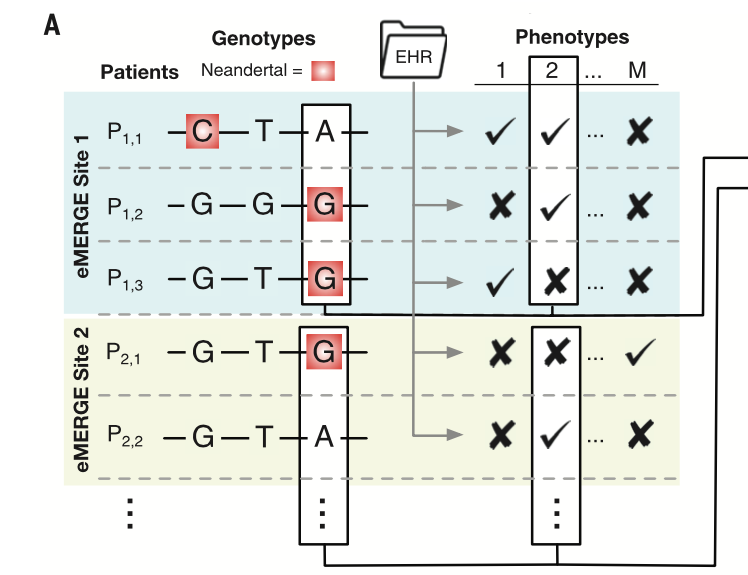
Displays the flow from genomic data to phenotypic data through EHRs when trying to determine the overall influence of Neandertal alleles together on traits in AMHs. The eMERGE Network was used to obtain EHRs containing genomics data for ~28,000 adults of European ancestry and data from the 1000 Genomes project to obtain genomic data for non European populations and did a comparison with the Altai Neadertal genome. The phenotypes were the derived based on EHR linked to genomic data. The figure displays the overall flow from how the phenotypes were obtained from patients’ genomic data using EHRs.
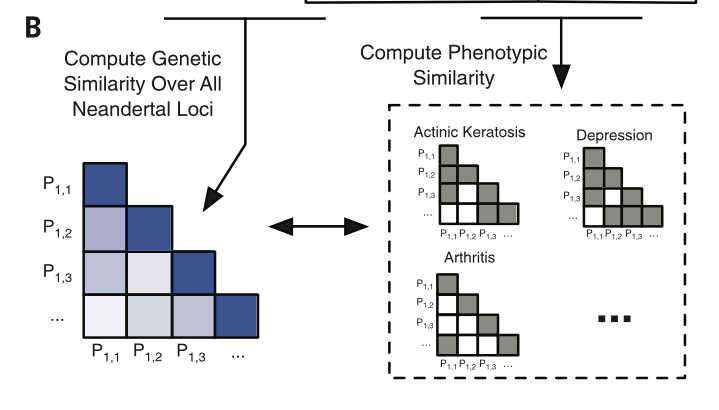
Displays how genetic similarity was found for of all pairs of individuals for 1495 using GTCA Neandertal loci and phenotypic similarity of 46 EHR-derived traits based on genomic enrichment. This method allowed the determination a general genotypic and phenotypic profile based on similarities (in these matrices) for a particular trait based on the various similarities of the sites among individuals.
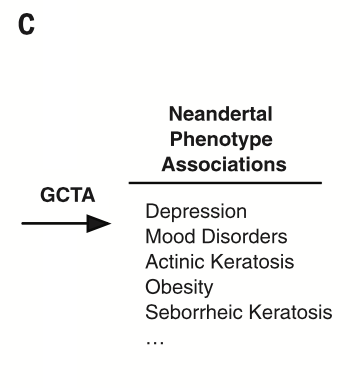
Displays a component of the work flow used to determine the overall influence of Neandertal alleles together on traits in AMHs. The group used mixed linear models in GTCA, which determine the significant contributions of the variants to the overall disease phenotypes in AMHs. From these data, the group derived a list of disease phenotypes associated with Neandertal SNPs and the overall risk contribution of Neandertal SNPs to these phenotypes.
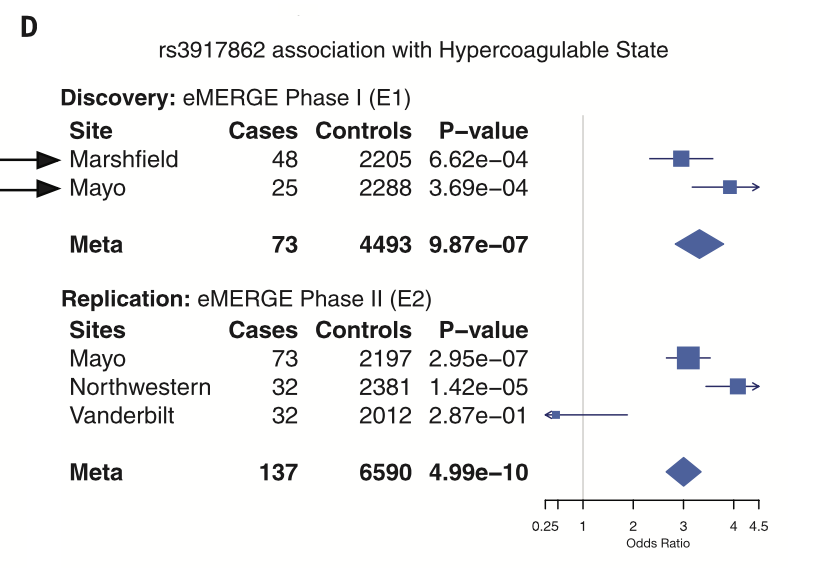
Discovery meta-analysis on both cohorts was used to identify (with replication) individual allelic associations using a PheWAS. This figure displays a break down for a particular SNP (rs3917862) with a Hypercoaguable state phenotype.Significant associations were found between specific SNPs and phenotypes based on the meta-analysis and these associations for rs3917862 are viewable in the figure. The chart breaks down the data sites and individual phenotypes for each and provides overall analysis (Each cohort is ordered from most to least significant).
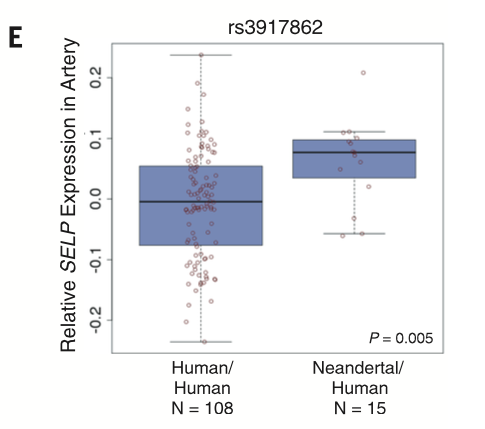
Displays that rs3917862 increased SELP expression using data from the Genotype-Tissue Expression (GTEx) Project. This provides further confirmation that the derived associations are valid and demonstrates that the Neadertal allele has a significant association with increased SELP. This method can be applied to the other SNPs as well.
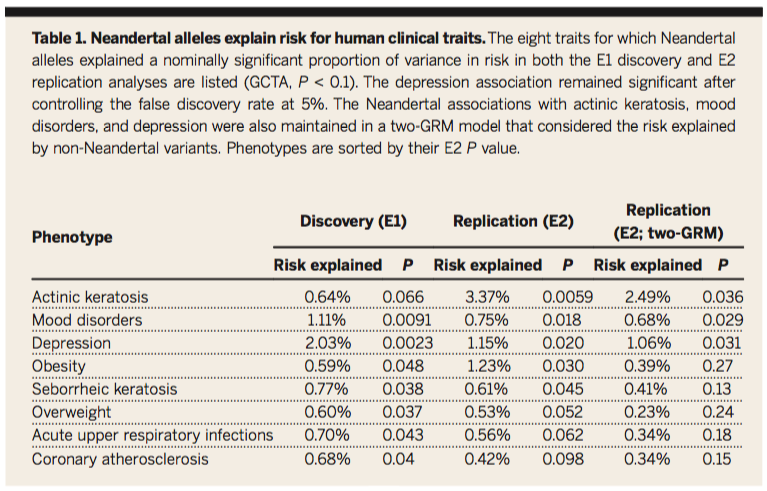
Table 1:
Displays the overall influences of these Neadertal alleles and the risk associated with disease phenotypes in a list generated by the mixed linear models and GTCA described in Figure 1C. A significant amount of risk associated with these various disease phenotypes can be explained by the overall presence of Neandertal alleles. These data provide us with a context for exploring areas of the genome that can tell us about the genetic admixture of Neandertal and AMH populations.
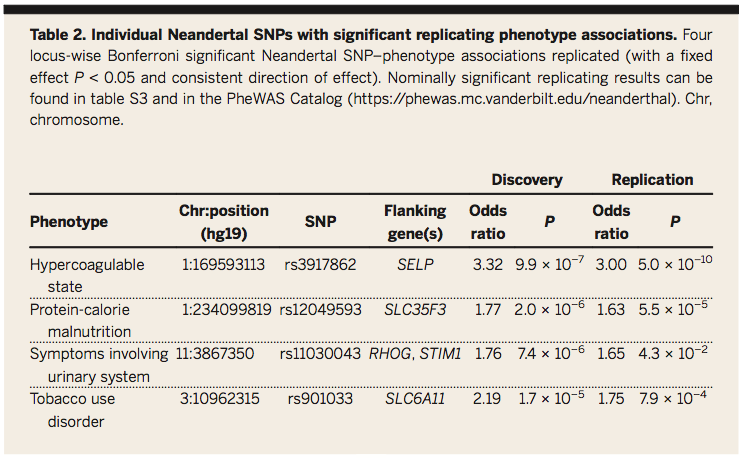
Table 2:
Displays specific SNPs have significant contributions to specific disease phenotypes. A meta analysis was performed on both cohorts and a locus-wise Bonferroni corrected significance threshold to identify significant Neandertal SNP-phenotype associations. Four specific Neandertal SNPs are identified that are clearly associated with particular disease phenotypes. Specific alleles are discerned and their individual contribution to risk determined. This list can inform theories as to why these diseases may appear in particular populations based on our understanding of the SNPs responsible and why these alleles may have been positively selected for in past populations. It also provides another layer of analysis for future studies.
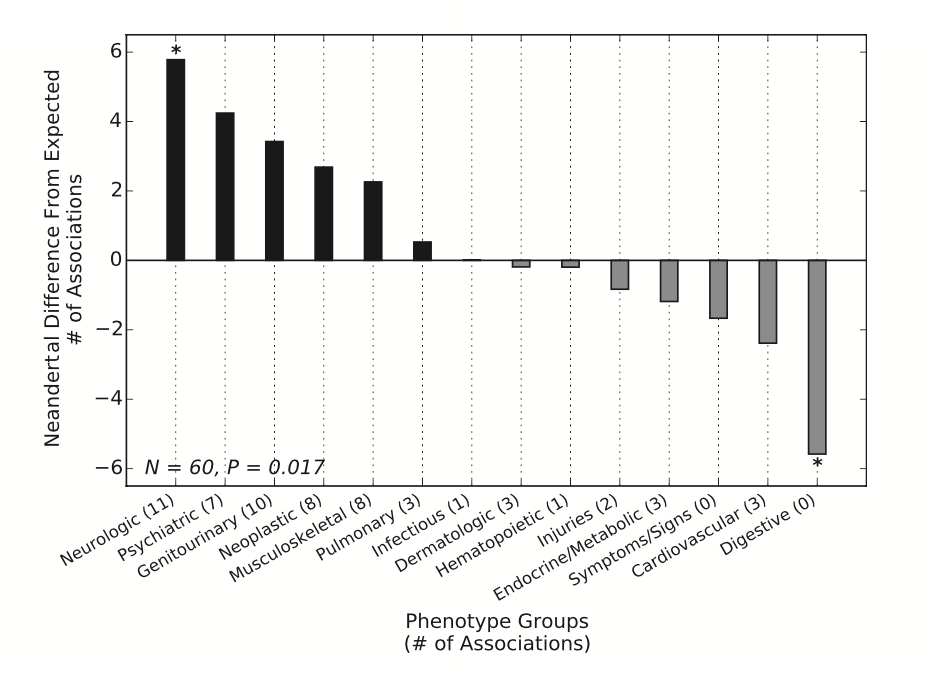
Figure 2:
Displays Neandertal SNPs enriched for association with specific classes of phenotypes using a comparison of distribution for replicating phenotype associations of a set of 1056 linkage disequilibrium (LD) pruned Neandertal SNP sets at a relaxed PheWas discovery threshold. The Neandertal SNPs significantly differ in the classes of phenotypes that they affect. These SNPs are associated with more neurological disorder phenotypes and fewer digestive phenotypes (far left and far right). Again, this informs our understanding of Neandertal and AMH genetic admixture, while also providing another layer of analysis that can be used in future studies.
References