This web page was produced as an assignment for an undergraduate course at Davidson College
Back to homepageIn "The Simons Genome Diversity Project: 300 Genomes from 142 Diverse Populations," Mallick et al. used Illumina sequencing to determine important elements in humanity’s evolutionary landscape, including measures of relatedness and differing rates of mutation between African and non-African populations, and the influence of early hominids on current populations. This paper is a response to the 1000 Genomes Project, which while analyzing a greater number of individuals, only looked at 26 geographically distinct human populations. The authors contend that the smaller populations that were overlooked in the 1000 Genomes Project are nonetheless important for understanding variation in modern humans, and intentionally chose subgroups to reflect “genetic, linguistic, and cultural variation”. (Mallick et al.)
The researchers sequenced 278 human genomes representing 142 human populations to 43-fold coverage using Illumina sequencing. After the application of filters to limit bias toward the reference genotype they amassed 34,300,000 SNPs and 2,100,000 indels. Giving weight to their theory that the 1000 Genomes Project provides an incomplete, they provide three examples of populations for which their data yielded greater heterozygosity: “11% in the KhoeSan and 5% in New Guineans and Australians.” (Mallick et al.) They further used FermiKit to genotype SNPs and indels, and lobSTR to genotype STRs.
Main Conclusions
· As in previous studies of humanity’s evolutionary landscape, they determined that sub-Saharan Africans retain the highest genetic diversity (measured as autosomal heterozygosity), which is consistent with the Out of Africa theory.
· Building on findings confirming that Neanderthal ancestry is present in all non-Africans, the researchers fine-tuned this using their data set. They determined that East Asians have the highest proportion of Neanderthal ancestry, and the South Asians tend to have a greater proportion of Denisovan ancestry than other Eurasians. However, some Australo-Melanesian populations retained over 5% Denisovan DNA, far outreaching the South Asian populations proportion of Denisovan ancestry.
· The common ancestral population to all modern human populations began to diverge around 200,000 years ago, but as all compared groups share “a substantial subset of their ancestors as recently as a hundred thousand years ago” this divergence was very slow. (Mallick et al.)
· While Australians, New Guineans, and Negrito populations do have both Neanderthal and Denisovan ancestry, introducing an earlier split between these populations and East Asians decreased the correlation of the model to the data. Therefore as with all other human populations, these populations are mainly descended from Homo sapiens with minimal contributions from Neanderthals and Denisovans.
· Non-Africans have significantly higher rates of mutation accumulation compared to sub-Saharan Africans.
· Despite the geographically widespread appearance of modern human behavior around 50,000 years ago, there was no selective genetic sweep throughout all human populations during this time. This indicates that the rapid changes to modern human behavior over the last 50,000 years were likely driven by environmental demands and cultural innovations.
Figure Interpretations
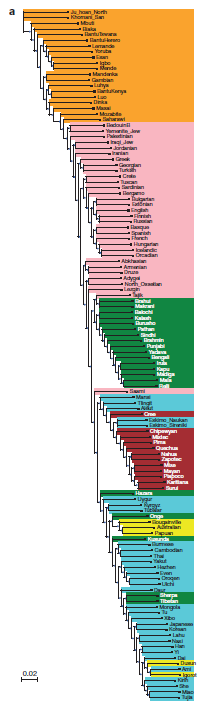
Fig 1a's phylogenetic tree was organized by determining the average divergence per nucleotide for each population pair and their fixation index. The color scheme indicated at the top of 1b indicates the region to which each population belongs.
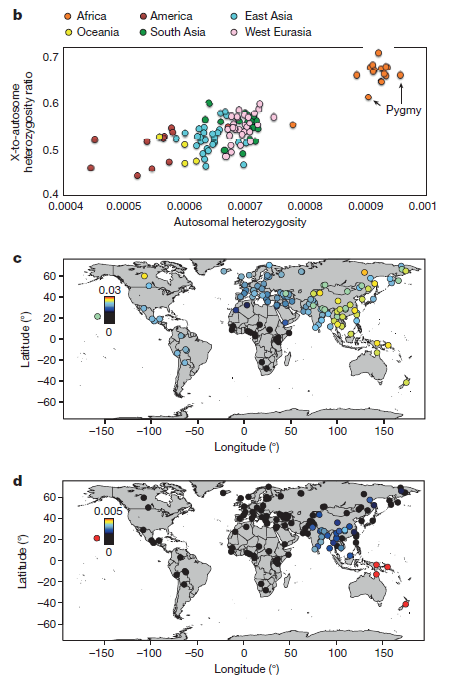
Figure 1b shows the average autosomal heterozygosity compared to the ratio of X-chromosome heterozygosity to autosomal heterozygosity for each population. All African populations have greater autosomal heterozygosity than all other populations, and have a higher average X-to-autosomal heterozygosity ratio.
Fig 1c is a heat map of Neanderthal ancestry in all sampled populations, showing that all non-sub Saharan African populations have maintained Neanderthal DNA, and that it is most prevalent in East Asian and Oceanic populations. Fig 1d is a heat map of Denisovan ancestry in all sampled populations, showing that it is present only in a subset of human populations mainly located in South Asia and Oceana.
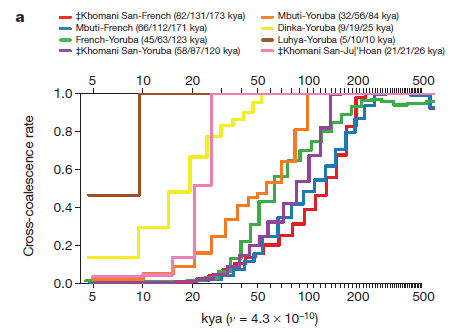
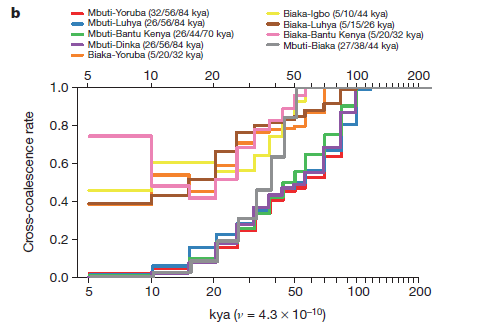
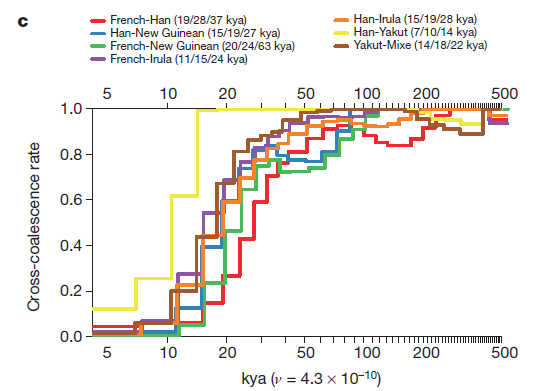
In Figures 2a, b and c the researchers model how many thousands of years ago two currently divergent populations shared a greater proportion of SNPs. They used four haplotypes to compare pairs of populations, using the pairwise sequential Markovian coalescent approach. Read from right to left, each of these graphs models a pattern of genetic divergence between pairs over time, with the estimated point at which 2 populations shared 25, 50, and 75% ancestry indicated to the right of the pair in each figure’s key.
2a compares selected African populations to each other and selected African populations with a French population, showing that the French population diverged from all African populations around 200,000 years ago (200 kya), and that African populations diverged from each other more recently.
2b compares selected African populations to each other, showing that cross-coalescence with Pygmy populations (Mbuti people) has steadily decreased from 100 – 10 kya, and that it has decreased at a faster rate than between other African groups. Additionally, this graph shows that from 17 – 5 kya the Biaka and Bantu Kenya populations increased in shared ancestry, probably indicating a gene flow event.
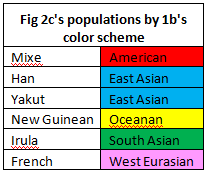
2c compares non-African populations from geographically diverse populations. Most populations began to diverge around 200 kya, and all populations had diverged by around 50 kya with the unsurprising exception of Han and Yakut populations. These are the only example of two populations from the same region portrayed in this graph. Additionally, while Han and Yakut populations diverged sharply around 15 kya, they maintain the highest proportion of cross-coalescence portrayed on this graph.
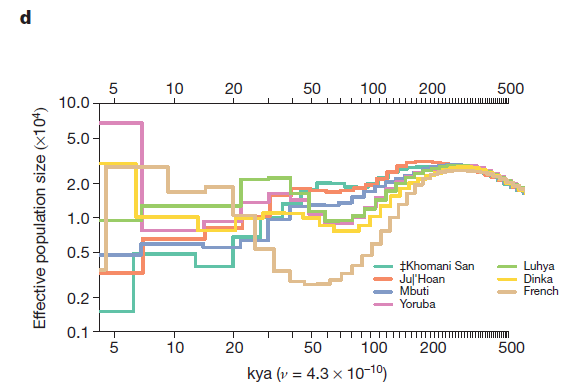
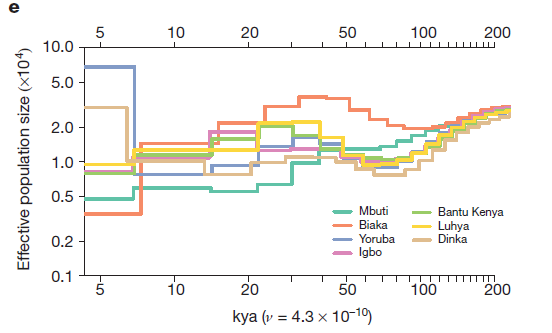
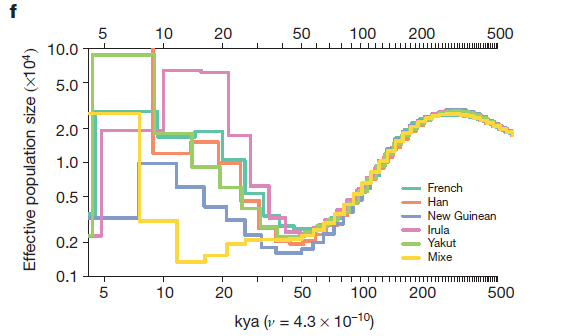
In figures 2d, 2e, and 2f the researchers model the effective population sizes of the populations used in 2a, 2b, and 2cs’ graphs, respectively. Effective population size is a measure of how many individuals’-worth of genetic data is present in a population, regardless of the actual number of individuals making up that population. It’s often used when assessing the viability of small and/or inbred populations, and in this case reflects the overall amount of genetic diversity of each population.
2d models selected African populations compared to the French population. Before 200 kya, the human population was restricted to sub-Saharan Africa and gene flow was abundant due to a lack of geographic barriers. The initial increase in effective population size from 500 kya to 200 kya for all groups probably indicates a slow accumulation of mutations and heterozygosity in this original population. But population divergence around 200 kya resulted in the founder effect, wherein any population founded by a subset of an original population will not maintain all of the genetic diversity of the original population, and will over-represent the genetic data of the founding population. This is particularly apparent in the steeper decline in effective population size of the French, who were more geographically removed from the original population and therefore could not regain genetic diversity through gene flow. The increase in effective population size (occurring in African populations around 80 kya and the French population around 50 kya) may represent an increase in actual population size and corresponding increase in heterozygosity, or a gene flow event.
2e models selected African populations. The concordance of effective population size is evident until almost 100 kya, and all maintain relatively similar effective population sizes, which is indicative of gene flow between populations.
2f models non-African populations. In the 150 kya following divergence from the original population all non-African populations decreased drastically in effective population size. This is a logical consequence of the founder effect, but these subpopulations were probably relatively small and completely isolated from the original population I believe that genetic drift also played a major part in decreasing their genetic variation. All except the Mixe population experiences a steady increase in effective population size from 50 to 10 kya, indicating that a slow accumulation of mutations was leading to greater variation within each population.
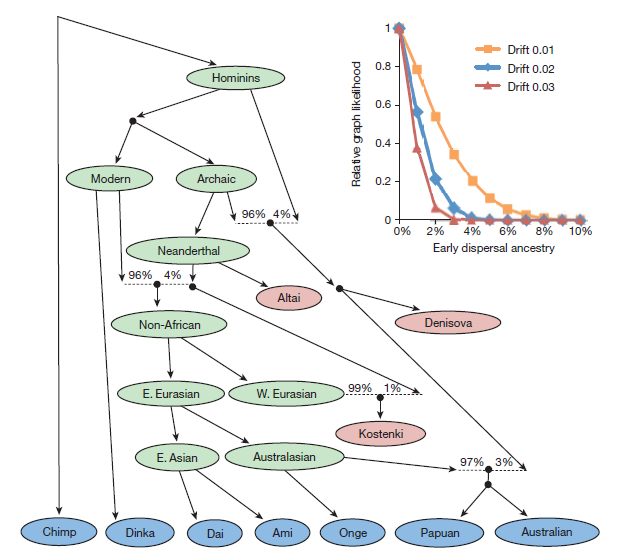
In figure 3 the researchers represent a graphical model of the degree of ancestral admixture with early hominids of six geographically diverse human populations. This chart was constructed through genetic analysis of samples of modern populations (blue), ancient populations (red) and inferred populations (green). Admixture events are represented by dotted lines, with Neanderthal DNA making up 4% of genetic variation in the original non-African population. The researchers indicate that modern Papuan and Australian populations share genetic variation with the extinct Denisovan population due to an admixture event with a population ancestral to the Denisovans, which contributed 3% to modern Papuan and Australian variation. This figure does not explain the presence of Denisovan ancestry outside of Oceana referenced in Fig 1d, particularly in South Asian populations.
The inset explains why the researchers ascribed such low percentages of ancient hominid ancestry to modern human populations. It describes the relative likelihood of modern human populations having 0-10% ancestry from earlier-dispersed hominids given the model of human dispersal presented in the main figure. They represent the timing of the split by indicating the amount of genetic drift, with smaller drift units corresponding to a more recent split. All drift conditions best match the model at 0% early dispersal ancestry. Additionally, the earliest split (drift 0.03) reaches a likelihood of 0 at around 3% early dispersal ancestry, as does the middle at 4% and the latest at 8%. The researcher therefore restricted early dispersal ancestry to “a few percent” to achieve thegreatest consistency both with their genetic data from modern populations and with their model. (Mallick et al.)
Analysis
While this paper is a logical rebuttal to the 1000 Genomes Project’s narrow geographic focus, this paper relies on an average sample size of of 2.11 individuals’ genomes per population. Li et al. demonstrate that there is vastly more variation within a population than distinctive variation between populations in “Worldwide Human Relationships Inferred from Genome-Wide Patterns of Variation”. This informs my main issue with the paper: A more robust sample size per population is necessary to determine statistically significant genetic differences between these populations. To this point, none of Mallick et al.’s figures indicate significant, and the paper often refers to ‘substantial’ rather than ‘significant’ results.
They also mention that “the true value of the human mutation rate…could plausibly be 30% higher or lower than the point estimate we use.” A large portion of this paper is devoted to dates and numbers (Fig 2a-f) dependent on this mutation rate, leading me to question the reliability of these models. Additionally, effective population size in figures 2d, 2e, and 2f are based on one diploid genome per population, and no explanation is given for the extreme changes in effective population occurring (in figures d, e, and f) in many populations from 5,000 to 15 kya. I am skeptical of the reliability of this information given they were extrapolating over hundreds of thousands of years based on only one individual’s genome.
Including a wider range of populations in genomic studies of human evolution is an important step forward in understanding our history and current evolutionary landscape, and over the course of the semester it’s become clear to me that science has historically ignored reality concerning minority populations. I appreciate that this paper endeavors to include a more robust sample of populations “genetic, linguistic, and cultural variation, ” and I believe that this approach is necessary for a veracious understanding of human evolution. (Mallick et al.) But Mallick et al. provides us no indication of how reliable their results are, and I would not stake my scientific credibility on their unknown p values.
References
Li, Jun Z. et al. “Worldwide Human Relationships Inferred from
Genome-Wide Patterns of Variation.” Science 319.5866 (2008):
1100–1104. Web. 27 Apr. 2017. Available from http://science.sciencemag.org/content/319/5866/1100
Mallick, Swapan et al. “The Simons Genome Diversity Project: 300
Genomes from 142 Diverse Populations.” Nature 538.7624 (2016):
201–206. Web. 27 Apr. 2017. Available from https://www.nature.com/nature/journal/v538/n7624/full/nature18964.html
Genomics Page
Biology Home Page
Email Questions or Comments: jaburtonakright@davidson.edu
© Copyright 2016 Department of Biology, Davidson College, Davidson, NC 28035