This web page was produced
as an assignment for an undergraduate course at Davidson College.
Helen Webster's Genomics
Home Page
DNA damage is a pervasive cause of sequencing errors, directly
confounding variant identification
Main Idea
To test the possibility that mutagenic damage causes sequencing inaccuracies
and variation, Chen et al. created a standard score (GIV score) for
determining read imbalances and sequencing variants. Public sequencing
datasets contain this mutant variation mixed in with natural variation, and
it is unclear to those who use the data that some variation is caused by
mutagens. Mutagenic damage of DNA causes an imbalance in base transversions
between the reads of the two DNA strands, and is a major confounding effect
of damage. This imbalance is used to the researchers' advantage, as it can
ideally be used as a basis to track mutagenic damage and therefore
sequencing errors in these databases, as opposed to natural variance present
in the population. This GIV score can ideally be used to accurately estimate
the damage present in public data sets. Chen et al. conclude that the GIV
score accurately quantifies mutagenic damage in somatic variant cells, which
occurs in very low frequencies. This affects a substantial portion of
My Opinion
I found this paper especially intriguing given the integral role DNA
sequencing has played in the biological research I have conducted at
Davidson, as well as vital to nearly every part of the work we read about in
our genomics course. It is incredible to me that mutagens could play such a
detrimental role not just physically for the DNA itself, but for further
findings and usaage of DNA reads and sequences. I think this paper did an
impressive job of establishing a necessary a score system for damage, that
would have obvious positive benefits for improving public data sets. In
addition, the display of proving the scoring system and proceeding to apply
it with tumor cells thoroughly convinced the reader of the benefit of the
GIV score in addition to the credibility of the method. Finally, this GIV
score is a novel idea with a necessary application, that I think can quickly
be useful to genomicists and bioinformaticists.
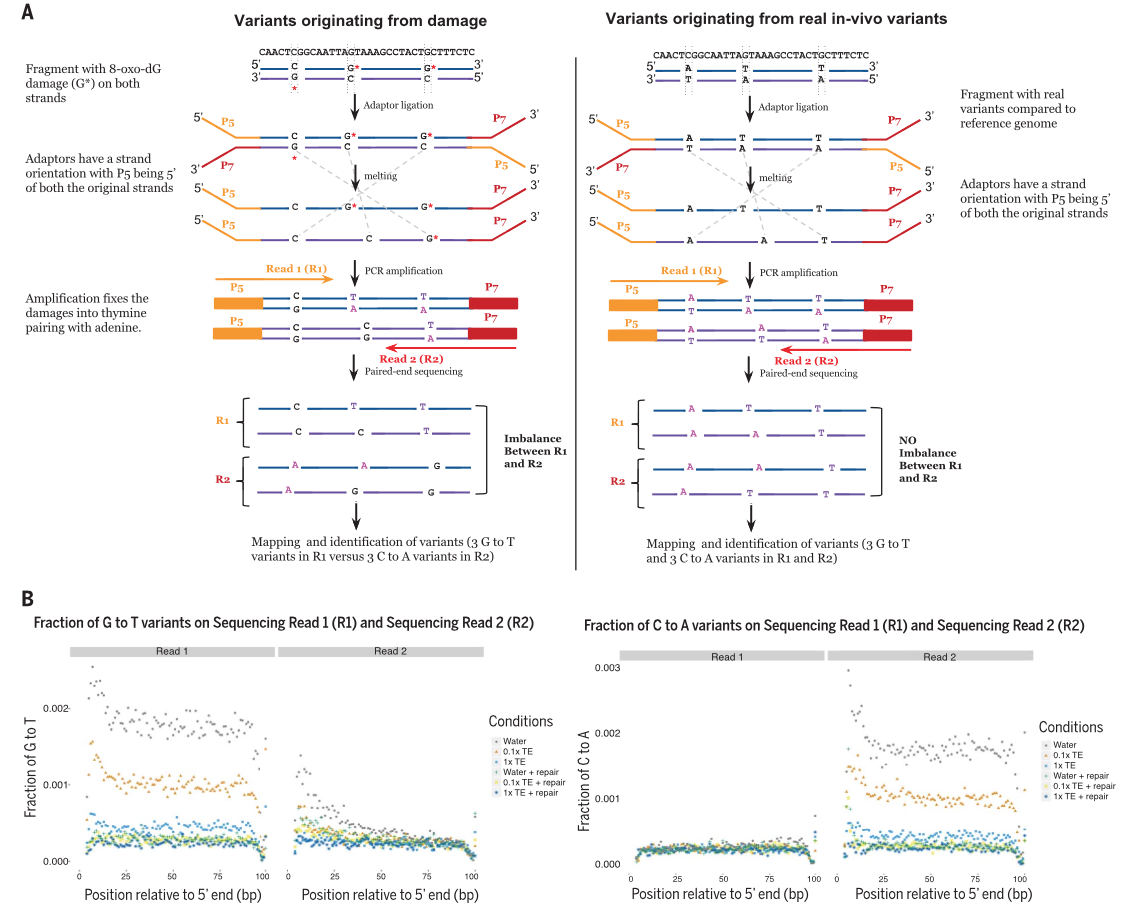
Figure 1
Figure 1A outlines the principle behind the
GIV score. Mutagenic variation causes an imblanace of transversion between
the two reads because the base switch between the two strands does not
happen equally. The left side of Panel A shows the base transversion
imbalance that results from sequencing mutagenically damaged sequences.
When the variant is just a natural SNP (right side of panel), reads from
both strands will transverse equally. The degree of imbalance due to
damage is the basis of the GIV score. Figure 1B visually shows the
fraction of G to T transversions and the inequity between reads 1 and 2.
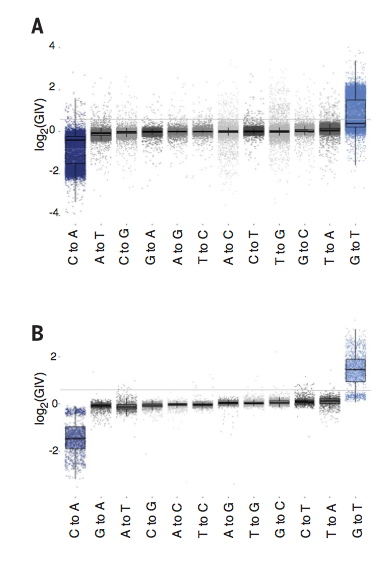
Figure 2
To estimate the amount of damage present in public DNA
sets, Chen et al. calculated the GIV scores for the 1000 Genomes Project
dataset (Figure 2A) and a subset of the TCGA data set (Figure 2B). The
gray line in both A and B demarcates a GIV score of 1.5, above which the
score implicates damage, below which is non-damage. Both sets show
widespread erroneous sequencing calls, 30% of which were G to T variant
reads. T to A and C to T were 0.5% and 3% of erroneous calls,
respectively. Overall, Figure 2 shows there is indeed DNA sequence
damage in public data sets that leads to erroneous sequencing calls in
at least 1/3 of G to T variant reads.
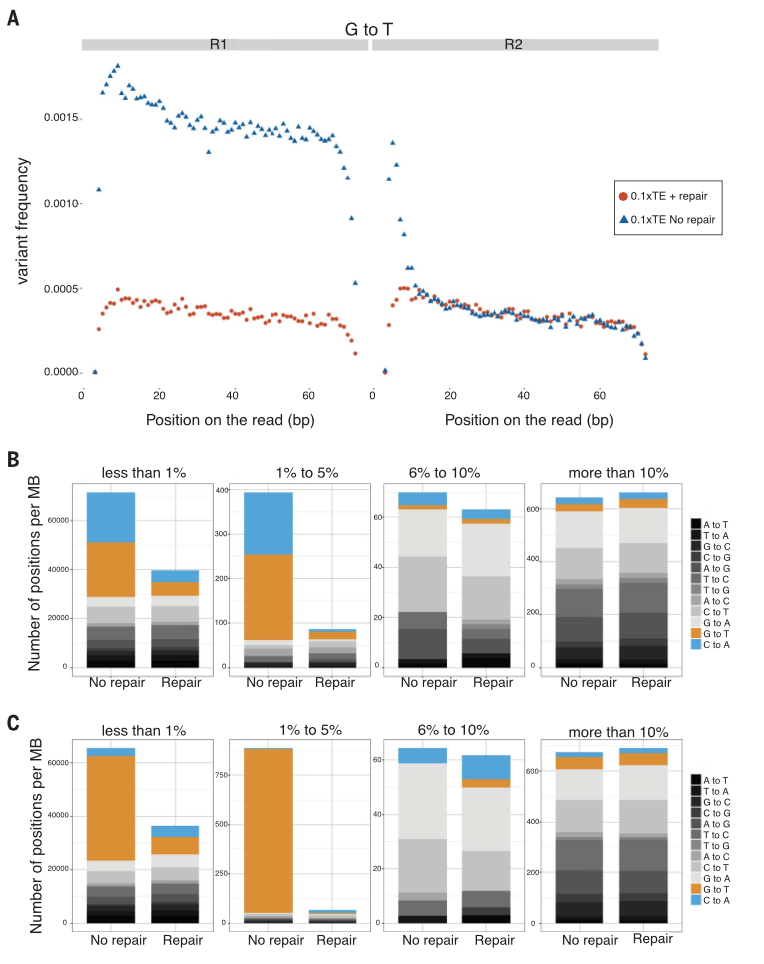
Figure 3
Supplementary data found G to T
transversions to be randomly generated, implying they occur at low
allelic fractions (known as somatic variants, as opposed to the high
frequency germline variants). Figure 3 looks at how damage affects
somatic variant identification. DNA repair eliminates 82% of G-to-T
and C-to-A variant positions in the low frequency groups (less than
1% and 1% to 5%), proving those positions are erroneous and due to
damage in somatic variants. This leads to false positives and direct
confounding in the identification of variance in sequences reads.
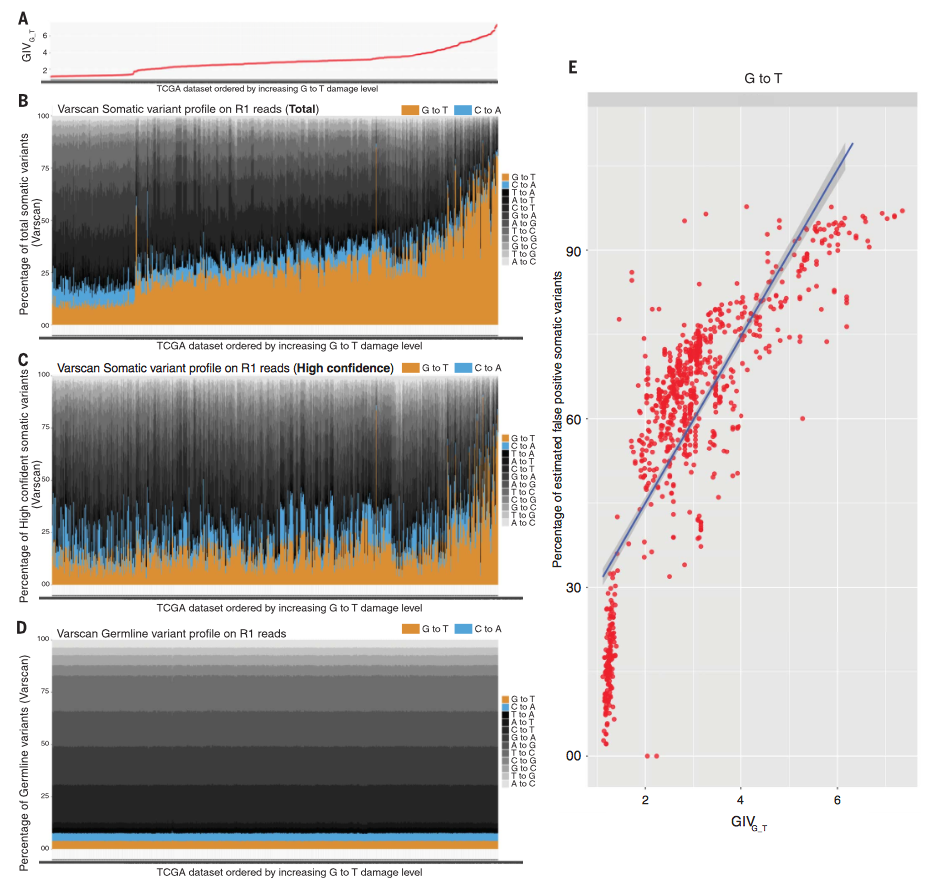
Figure 4
Figure 4 sorts approximately 1800 tumor sequencing runs by
G-to-T variant GIV score. There are more G-to-T somatic variants than
C-to-A, and the fraction increases with increasing GIV score. In addition,
panel 4D shows germline variants remaining consistent in GIV score.
Estimated false positives in somatic variants is strongly correlated to
estimated damage in these tumor samples, ultimately supporting the
application of the GIV score to accurately detect high somatic damage and
false positives in sequence reads.
Citation: Chen L, Liu P, Evans T,
Ettwiller L. (2017). DNA damage is a pervasive cause of sequencing errors,
directly confounding variant identification. Science 355, 752-756.
Helen's Home Page
Genomics
Page
Biology
Home Page
Email Questions or Comments: hewebster@davidson.edu
© Copyright 2016 Department of Biology,
Davidson College, Davidson, NC 28035