The Purification and Characterization of Glucose-6-Phosphate Dehydrogenase fromChlamydomonas reinhardtii
Bradley S. Ellison
Honors Thesis
1997
ABSTRACT
Glucose-6-Phosphate-Dehydrogenase (G6PD) is an NADP+-dependent enzyme essential to the oxidative pentose-phosphate cycle. Experimental parameters were designed to quantitatively purify and characterize G6PD from Chlamydomonas reinhardtii, an important biological model system. G6PD was purified to apparent homogeneity using Amicon Matrex Gel Blue A. Electrophoretic analysis suggests only one functional species of G6PD exists for C. reinhardtii with an approximate molecular size of 180 kD, indicating a tetrameric configuration. Km values for DL- G6P and NADP+ were 103.0Å10.5 x 10-6 M and 4.8Å0.153 x 10-6 M, respectively. The activity of the enzyme appears to be enhanced in the presence of Mg2+, Mn2+, and Ca2+. However, G6PD cannot utilize Cu2+, Sr2+, or Zn2+ ions as electron acceptors. Moreover, Cd2+, Hg2+, and Ni2+ appear toxic to enzyme activity. G6PD has a broad pH optimum of 7.6 to 8.3 and is stable to a maximum temperature of 60 %C.
INTRODUCTION
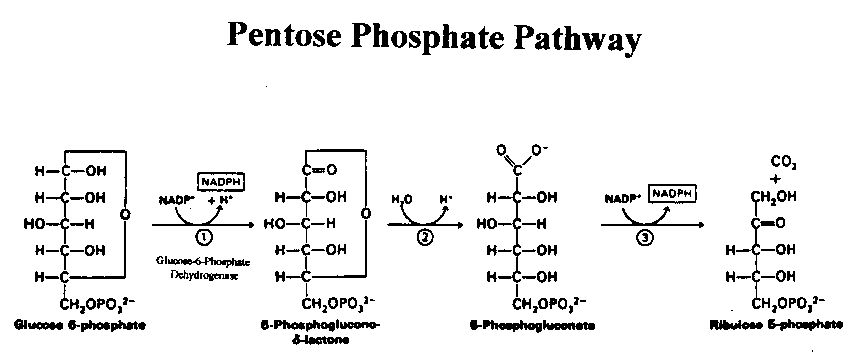
Enzymes are biochemical catalysts that have highly distinctive and biologically crucial properties. An individual enzyme usually catalyzes a single reaction or a set of closely related reactions, whereby the energy of the reactants is converted into an altered form of energy with high efficiency. Glucose-6-phosphate dehydrogenase (G6PD; E.C. 1.1.1.49) is a ubiquitous enzyme that catalyzes the initial step in the pentose phosphate pathway and regulates its oxidative branch (Figure 1).
Figure 1: 2 NADPH are produced in the conversion of Glucose-6-Phosphate into Ribulose 5-Phosphate
In eukaryotes, G6PD is NADP+-dependent and essential in the formation of pentose, the precursor to nucleic acids and nucleotide coenzymes. G6PD was initially described in 1931 by Warburg and Christian in horse erythrocytes, and has since been studied from many varied sources (Arese and Flora, 1990).
Considered ancient in evolution, G6PD is found in every tissue of all contemporary organisms, suggesting G6PD acts as an important housekeeping enzyme critical to the maintenance of homeostasis. The enzyme is highly specific for the reaction where glucose-6-phosphate (G6P) is oxidized to 6-phosphoglucono--lactone. In the course of this reaction, NADP+ is reduced to NADPH, which serves as an integral component in many biosynthetic and detoxification processes. In addition to NADPH, the pentose phosphate pathway also produces ribulose-5-phosphate, which can be converted to other sugars via the "Pentose Phosphate Carbon Shuffle."
The unicellular green alga, Chlamydomonas reinhardtii, has emerged as a powerful molecular and genetic model system, although relatively little is known about its biochemical mechanisms (Harris, 1989). In many cases, single-celled eukaryotes contain the basic elements characteristic of more complex organisms inrelated phyla. Common structural motifs demonstrate how the differences in genetic organization of individual genes and proteins are nearly indistinguishable, even across an assortment of species traditionally considered evolutionarily diverse. Recently, several NADP+-dependent dehydrogenases from Chlamydomonas have been partially purified and characterized (Adams, et al. 1995). However, procedures that have been successful in other organisms (Geer, et al. 1980; Williamson, et al. 1980 a, b) have not been as facile using Chlamydomonas as the source organism, implying these enzymes from Chlamydomonas may be different in some subtle manner(s) affecting protein-binding affinities.
Dye-ligand chromatography is a form of affinity chromatography, in which synthetic textile dyes are used in lieu of natural substrates, cofactors or effectors commonly employed as immobilized ligands. Since the introduction of the technique using Cibacron Blue 3GA dye for the purification of nucleotide coenzymes, dye-ligand chromatography has greatly expanded to include the purification of nearly two hundred enzymes and other proteins (Amicon, 1989). As an NADP+-dependent dehydrogenase, G6PD from Chlamydomonas appears to be a logical candidate for purification using dye-ligand chromatography.
Once G6PD is quantitatively purified, an extensive characterization of the enzyme can be achieved that would afford special insight into the intrinsic properties of G6PD from primitive eukaryotes. The characterization explores enzymatic properties related to pH and temperature optima, substrate kinetics, divalent metal requirements, subunit molecular weight, as well as the number of functional species of G6PD in the organism. This research will provide comparative information on the similarities and differences between G6PD from an assortment of evolutionarily diverse systems such as bakers yeast, maize, Drosophila, humans and Chlamydomonas.
BACKGROUND
Enzyme Specificity
By reducing activation energy, enzymes accelerate biochemical reactions approximately a million-fold, suggesting that most reactions within biological systems do not occur at perceptible rates in the absence of enzymes. The degree of enzymatic specificity, both in the reaction catalyzed and in their choice of substrates, is usually high and sometimes absolute (Stryer, 1995). Substrates bind to a specific region on the enzyme called the active site. Indeed, the catalytic specificity of enzymes depends in large part on the specificity of the enzyme-substrate binding mechanism. Comprising only a small portion of the total volume of an enzyme, the active sites are precisely defined by the arrangement of amino acids contained within this point of substrate interaction.
Pentose Phosphate Pathway
The pentose phosphate pathway is also known as the hexose-monophosphate shunt and involves three major steps (Figure 1). In the first step, G6PD oxidizes glucose-6-phosphate to 6-phosphoglucono--lactone. In the process, NADP+ is reduced to NADPH. Next, lactonase converts the 6-phosphoglucono--lactone to6-phosphogluconate in the presence of water. Finally, 6-phosphogluconate is converted to D-ribulose 5-phosphate by 6-phosphogluconate dehydrogenase (6PGD). Thus, the primary end-products of the pathway are ribulose 5-phosphate and two molecules of NADPH. In fact, the pentose phosphate pathway is the major source of NADPH, a biochemical vitally important in metabolism because of its reducing power essential for the production of fatty acids and steroids (Luzatto and Battstuzzi, 1985).
Ribulose-5-phosphate may be transformed into nucleotides used in the synthesis of DNA or RNA. However, not all ribulose-5-phosphate produced will go to produce nucleotides. In some cases, ribulose-5-phosphate is processed by the non-oxidative part of the pentose phosphate pathway, commonly known as the "pentose phosphate carbon shuffle." As the name implies, this pathway cleaves and rearranges ribulose-5-phosphate to generate sugars containing three to seven carbons. A primary driving force for the carbon shuffle is the generation of triose and hexose sugars, such as 3-phosphoglyceraldehyde and fructose-6-phosphate, which serve as intermediates in glycolysis. Thus, the non-oxidative part of the pathway not only prevents the accumulation of excess ribulose-5-phosphate, but also links the pentose phosphate pathway to glycolysis.
G6PD
In humans and Drosophila, the gene encoding G6PD is located on the X- chromosome, thereby subjecting the enzyme to phenomena like dosage compensation and X-inactivation. At present, the genetic organization for Chlamydomonas is not fully understood. The genome has not been thoroughly mapped and is currently comprised of only linkage maps; however, the G6PD gene has yet to be located (Harris, 1997). Multiple G6PD isoforms have been identified in humans, Drosophila, and related species of plants, suggesting that different forms of the enzyme may be present at certain times and locations to accommodate an organism's specific biochemical requirements (Vulliamy, et al. 1992; Williamson, 1986; Klein, 1984). For many species of plants, the enzyme has two classifications, either chloroplastic or cystolic (Fickenscher, 1986). As of yet, only one distinct form of G6PD has been identified for Chlamydomonas and it is principally chloroplastic, although the existence of a second isoform lies within the realm of probability. Previous studies have shown that redox activation states of G6PD may change relative to the photosynthetic electron supply (Klein, 1984; Farr, 1994). Also, G6PD activity has been discovered to adapt to nitrogen-limiting environments, indicating G6PD may be associated with mechanisms involved in the survival of cells with limited nitrogen resources, with the optimization of nitrogen acquisition when resources become available (Huppe and Turpin, 1996). Hence, collected data suggests G6PD may very well have a second isoform, yet, no experiment has conclusively explored the isolation and distinction of G6PD variants from Chlamydomonas.
The functional G6PD protein is normally an oligomeric molecule composed of identical subunits. Monomeric subunits aggregate forming the oligomeric enzyme. The molecular weight of the subunit is lower, on the average, in bacteria and in lower prokaryotes than in mammals (Luzzatto and Battistuzzi). Invariably, the smallest oligomeric structure with catalytic activity is the dimer (Luzzatto and Battistuzzi). Inmost species, the G6PD subunit has approximately 500 amino acid residues with a molecular weight on the order of 50-60 kDa (Luzzatto and Battistuzzi). In more primitive organisms, for example the gram-positive bacterium, Leuconostoc mesenteroides, the G6PD subunit contains 485 residues, while contrasted with human G6PD, which contains 515 residues per subunit (Rowland, et al. 1996; Takizawa, et al. 1986).
Although uncertainty surrounds the binding order in the NADP+ reaction of G6PD, there is evidence suggesting NADP+ binds first, inducing a conformational change in the assembly that promotes the subsequent binding of G6P (Crans amd Schelble, 1990). Once G6P is securely bound in the substrate active site, it is oxidized and then released (Figure 3).
As G6P is being oxidized, the coenzyme NADP+ is reduced to NADPH, which is then released. The NADP+-linked G6PD reaction mechanism is apparently quite specific, as NADP+ always binds before G6P, and NADPH is always released last (Crans and Schelble, 1990).
The interrelatedness of G6PD from diverse species has been shown through the structural analysis of G6PD from Pichia jadinii, where this species of yeast demonstrated 32-37% residue identity to selected bacterial G6PD, 47-48% to the mammalian and fruit fly enzymes, and 62% to baker's yeast enzyme (Jeffrey, et al. 1993). The striking similarities between heterogenous glucose-6-phosphate dehydrogenases suggest that the enzyme is structurally characterized as a homologous protein. In fact, the trend in homology may be further applied to the study of human G6PD, offering significant insight into human microevolution, as well as the widespread disease, G6PD deficiency.
The biological importance of G6PD is vindicated through its conservation maintained by evolution, whereby all organisms exhibit G6PD activity, albeit at varying levels (Luzzatto and Battistuzi, 1985). The study of G6PD has gained momentum due to the pathophysiology associated with human G6PD deficiency, which affects over 400 million people worldwide (Vulliamy et al., 1992). The erythroenzymopathic condition occurs when an affected individual becomes exposed to certain endogenous or exogenous toxic oxidants, such as primaquine or fava beans and experiences an episode of hemolytic anemia.
As a line of defense, red blood cells (RBC) possess a simplified mechanism designed to accomplish necessary tasks and to defend the functional properties from chemical and physical aggressors (Arese et al., 1990). Oxidation is harmful to all RBC constituents, but sulfhydryl groups are particularly at risk. A large number of biochemical functions depend critically on a small number of essential thiols. To counteract thiol oxidation, RBCs almost exclusively uses reduced glutathione. Reduced glutathione is necessary for the maintenance of sulfhydryl groups within RBCs and RBC membranes. Since oxidized glutathione is reduced by NADPH, and since the first and main source of NADPH is the reaction catalyzed by G6PD, this enzyme occupies a central position in the assurance of the stability and viability for RBCs. When the major source of NADPH is deficient, the oxidative stress in time leads to extensive cross-linkages of membrane proteins, resulting in RBCs that are completely rigid . Unable to flow through the narrow dimensions of the vascular network, affected RBCs become physically trapped within blood vessels, only tosuffer eventual phagocytic destruction (Arese et al., 1990).
Affinity Chromatography
The technique of affinity chromatography has become a well-accepted and widely useful tool in the purification of enzymes and other proteins. Unlike other commonly used fractionation methods (salt precipitation, gel filtration, ion exchange, and electrophoresis) which separate molecules according to nonspecific characteristics (solubility, charge, molecular weight, or electrophoretic mobility), affinity chromatography makes use of the highly specific binding sites usually present in biological macromolecules, separating molecules on their ability to bind a particular ligand. Covalent bonds attach the ligand to an insoluble, porous support medium in a manner that overtly presents the ligand to the protein sample, thereby using natural biospecific binding of one molecular species to separate and purify a second species from a mixture.
Affinity chromatography purifies proteins in a four stage process (Figure 4).
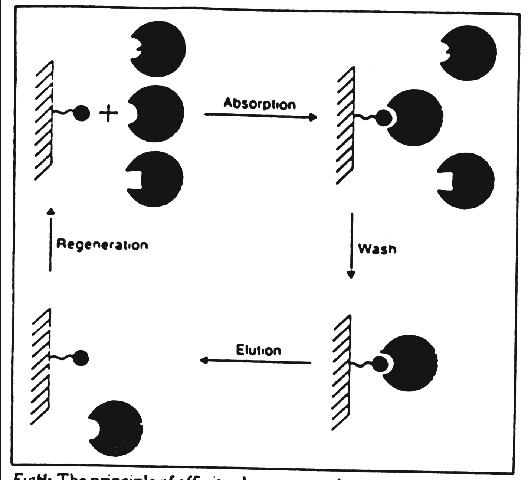
Figure 4: The principle of affinity chromatography.
In the first stage, known as the "Adsorption phase," the target protein binds to the ligand-column complex. Next, the "washing phase" uses an equilibration buffer to remove impurities and unbound material. The third phase, known as the "Elution phase," extracts bound molecules from the column. Finally, in the "Regeneration phase," the column is cleaned of excess bound material. The remaining step simply includes a functional assay to locate the greatest concentration of the targeted enzyme, which at that point can be concentrated further, in order to perform a characterizational analysis of the enzyme.
Most work with affinity chromatography has been done with the Cibacron Blue 3GA ligand. The Matex Blue A ligand is a slight variation of Cibacron Blue 3GA. Matrex Blue A is coupled directly to an agarose support through the triazine ring by CNBr (Figure 5).
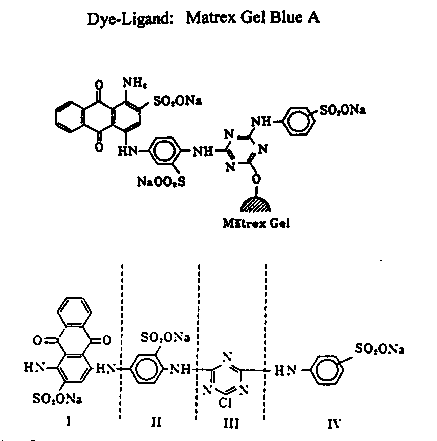
Figure 5: Four structural components of the blue
A dye-ligand: I- sulfonated anthraquinone (chromophore), II- sulfonated
benzene (bridge), III- triazine ring (active chlorine shown in agarose binding
site), IV- sulfonated benzene (terminal)
Although many enzyme purifications have been achieved with Cibacron Blue 3GA, there is no way to predict a priori which, if any, ligand is best for a particular protein from any organism. Amicon Matrex Blue A contains amino groups that are employed as the ligand interface for NADP-dependent dehydrogenases (Amicon, 1993).
Theories of dye-protein interaction have focused on the fact that the dye interacts specifically with a particular structural feature of most dehydrogenases and other enzymes at the "dinucleotide fold" (Amicon, 1993). According to the dinucleotide fold theory, enzymes containing this feature bind to Blue A columns, while enzymes and other proteins not containing the fold flow on through the column. Thus, the Blue A molecule serves as a selective probe for this structural feature in proteins. The dinucleotide fold consists of 150-residue polypeptide chain arranged in four to six parallel beta-strands connected by several alpha-helical strands about the beta-sheets (Amicon, 1993). This structure is found in a wide variety of dehydrogenases and several other enzymes and is remarkably well preserved in evolution, which suggests G6PD may contain a dinucleotide fold.
Methods and Materials
Chemicals and Reagents
All routine chemicals and buffer components were obtained from Sigma Chemical Company, except the ammonium sulfate used specifically for protein precipitation, which was obtained from Fisher Scientific Company. Matrex Gel Blue A was obtained from Amicon Division, W.R. Grace & Company. Sephacryl S-300 was obtained from Pharmacia Fine Chemicals. Electrophoretic materials were obtained from BIO-RAD, which included a Mini-PROTEAN II electrophoresis cell and BIO-RAD pre-stained molecular weight standards (Myosin, 199 kDa; -Galactosidase, 120 kDa).
Biological Materials
A wild-type strain of C. reinhardtii (cc125) was obtained from Dr. E. Harris, Chlamydomonas Genetics Center, Botany Department, Duke University, Durham, NC. Cells were inoculated from TAP (tris-acetate phosphate) plates into 2 L flasks containing liquid TAP medium (Harris, 1989). Cultures were grown for five days with continuous light at 23 - 26 %C without agitation or bubbling. Cells were harvested by centrifugation at 17,000 g for 5 min, resuspended in TEM buffer (0.1 M Tris, 5 mM EDTA, 1 mM 2-mercaptoethanol, pH 7.6) and frozen at -70 %C.
Ammonium Sulfate Precipitation
When needed, suspensions of Chlamydomonas cells were thawed slowly, allowing ice crystals to rupture cells and organelles. The resultant slurry was centrifuged at 17,000 g for 15 min to remove cellular debris. One milliliter of the supernatant was labeled as the crude homogenate, while the remaining supernatant from centrifugation was brought to 30% saturation with solid ammonium sulfate and stirred for 30 min at 4 %C, then centrifuged at 17,000 g for 20 min. The resultant supernatant was brought to 55% saturation with solid ammonium sulfate, stirred for 30 min and centrifuged at 17,000 g for 20 min. The pellet was resuspended in a minimum volume (4 - 8 mL) of 20 mM Tris/HCl buffer, pH 7.6, and pumped onto Matrex Gel Blue A (1.6 X 25 cm), previously equilibrated with 20 mM Tris buffer. The column was then washed with 100 mL of 20 mM Tris buffer. G6PD was eluted from the column using 1.5 M KCl in 20 mM Tris buffer, pH 7.6. All purification steps were performed at 0 - 4 %C. Fractions containing G6PD activity were identified by assaying 100 &l aliquots for G6PD activity. Fractions containing significant activity were pooled and concentrated using an Amicon filtration unit fitted with XM50 filters and applied to a Sephacryl S-300 column (2.6 X 85 cm). The protein was eluted with TEM buffer containing 0.05 mM NADP+ to stabilize the enzyme. Fractions containing G6PD activity were concentrated and used to determine parameters of G6PD activity.
Protein Assays
Protein concentrations were determined according to the method of Lowry, et al. (1951). Triplicate assays were performed for each sample, using BSA as the protein standard.
Enzyme Assays
NADP+-G6PD activity was measured by monitoring the reduction of NADP+ at340 nm in 1 mL assays containing 0.21 M Tris, 0.144 mM NADP+, 2.64 mM MgSO4 and 1.4 mM 6-phosphogluconate with a Beckman Model 35 spectrophotometer equipped with a heated cuvette chamber and water circulator set at 30 %C. Routinely, 10 &L aliquots of enzyme preparation obtained from the Matrex Blue A column were used to initiate reactions.
pH Optimum
The pH optimum of G6PD activity was determined by varying the pH of the assay buffer solution, while holding all other parameters constant. Triplicate assays were performed at each pH tested.
Temperature Optimum
The temperature optimum of G6PD activity was determined by varying the temperature at which assays were performed, while holding all other parameters constant. Triplicate assays were performed at each experimental temperature.
Molecular Size
Estimations regarding the degree of purity for the native enzyme, as well as subunit molecular sizes of G6PD, was obtained using an 8% polyacrylamide gel were obtained using a BIO-RAD Mini-PROTEAN II electrophoresis cell with pre-stained molecular weight standards (For standards see Chemicals and Reagents).
Substrate Kinetics
The kinetics for each substrate were determined by measuring the reduction of NADP+ at 340 nm and 30 %C at seven concentrations of substrate, while holding all other assay conditions constant. Triplicate assays were performed at each concentration tested. Values of Km were determined by double reciprocal plot regression analysis (Lineweaver-Burke Plot).
Divalent Metals
To determine if G6PD has an absolute requirement for a divalent ion and if metal ions other than Mn2+ and Mg2+ could serve as electron acceptors, assay solutions were prepared without added metals. Enzyme activity assays were carried out under otherwise standard conditions. After two minutes of reaction, 10 &L of concentrated ion solution were added to the cuvette to a final concentration of 2 mM. The reaction then continued for an additional two minutes.
RESULTS and DISCUSSION
Matrex Blue A Selection
Five different dye-ligand matrices were tested to measure their binding affinities for G6PD from baker_s yeast, Sarchomyces cerevisie (type VII: Sigma # G-7877). The five Matrex Gel dye ligands were: Blue A, Red A, Orange A, Green A and Blue B (Figure 6).
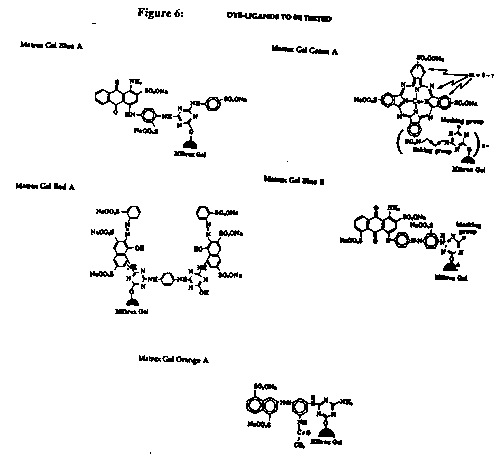
Figure 6: Dye-ligands to be tested.
Each of the five columns was washed with 20 mL of 20 mM Tris, pH 7.45. The commercial enzyme preparation was diluted 1:100 (10 &l of G6PD in 1 mL of Tris assay buffer with NADP+) and 100 &l were placed on each dye-ligand column. Each dye-ligand matrex was next washed with 20 mM Tris, pH 7.45, collecting approximately 1 mL fractions. Once four 1 ml fractions from each dye-ligand matrex were obtained, 4 mL of 1.5 M KCl in 20 mM Tris buffer, pH 7.45 were pumped over each matrex dye-ligand. From each 1 mL fraction, aliquots of 100 &l were assayed for G6PD activity.
These tests demonstrated that Matrex Blue A binds G6PD more effectively than the other dye-ligands (Table 1). Matrex dye-ligand Blue A quantitatively bound and released G6PD and was therefore chosen as the best dye-ligand to purify G6PD from Chlamydomonas reinhardtii.
Matrex Blue A Selection
Matrex Dye- Ligand |
Blue A |
Red A |
Green A |
Blue B |
Orange A |
Percentage (%) of Protein Recovered |
181 |
113 |
109 |
73 |
35 |
Table 1:
Percentage of G6PD activity obtained from five different Matrex gel dye-ligands,
using
Sarchomyces cerevisie as as the source organism.
Purification of G6PD
The 30-55% ammonium sulfate pellet from Chlamydomonas reinhardtii was loaded onto the Matrex gel Blue A column. The column was then washed with 60 mL of 20 mM Tris, followed by 60 mL of 1.5 M KCl. The column was then washed with 50-100 mL of 20 mM Tris. When the eluent was assayed for G6PD activity, the intense peak of activity was identified about 14 fractions after the 1.5 M KCl-20 mM Tris solution was applied to the column. The four fractions containing the most activity were consolidated and concentrated to an approximate volume of 1 mL. The consolidated fractions represent approximately a 100-fold purification of G6PD from Chlamydomonas. The concentrated pooled fractions served as the source of G6PD for the characterizational analysis of the enzyme.
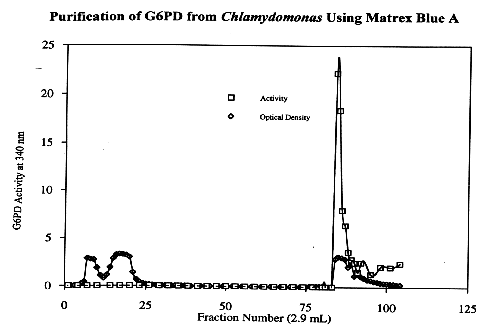
Substrate Kinetics
Interpretation of the reciprocal plot regression using seven incremental variants of G6P concentration indicated the Km of G6P for G6PD to be 103.0_10.5 &M. Km for G6P from human G6PD is 50-70 &M, while Km for G6P from Drosophila G6PD is 200-300 &M and 120 uM from pea leaves (Table 2; Luzzatto and Battistuzzi).

Table 2: Kinetic survey obtained with triplicate
enzyme assays for the two substrates
for G6PD from Chlamydomonas reinhardii
Therefore, the Km value for G6P obtained with G6PD from Chlamydomonas falls in the range of published values from other organisms. When presented with 2-deoxy G6P and G6P, G6PD demonstrated a preference for G6P, however also exhibited 2-deoxy G6P may be used as a substrate as much as 10%. However, 2-deoxy-G6P does not compete or interfere with G6P at the substrate active site (Data not shown).
As an NADP+-dependent dehydrogenase, G6PD uses NADP+ as a second substrate. The respective Km for NADP+ for G6PD was determined to be 4.8_ 0.153 &M. The Km for NADP+ from human G6PD is 2.9-4.4 &M, while Km from Drosophila is 1.1-5.8 &M and the Km is 13.5 &M in pea leaves (Luzzatto and Battistuzzi). Again, the results include a Km value for NADP+ from G6PD in Chlamydomonas to be within the range of other kinetic values for the coenzyme.
As the secondary substrate in G6PD, NADP+ acts to stabilize the enzyme as an oligomer and enhance the catalytic propensity (Rowland, et al., 1994). The smaller Km value for NADP+, when compared with the Km value for G6P, reflects the catalytic necessity of NADP+, however, also depicts the primary function of G6PD to be the oxidation of G6P to phosphoglucono-_-lactone. The most striking trend when comparing bacteria, lower eukaryotes, and mammals is the decrease in Km for both G6P and NADP+, suggesting that the fit of these ligands has been perfected, while the affinity for the product-inhibitor NADPH changes much less (Luzzatto and Battistuzzi).
Molecular Features of G6PD and Evolution
SOURCE |
Average Molecular Weight of Monomer |
Dimer |
Tetramer |
Multimer |
Average Km for G6P (uM) |
Average Km for NADP+ (uM) |
Bacteria |
56,800 |
+ |
+ |
+ |
1500 |
55 |
Lower Eukaryotes |
54,700 |
+ |
+ |
- |
212 |
30 |
Chlamydomonas |
45,000 |
- |
+ |
- |
103 |
4.8 |
Plants |
55,000 |
+ |
+ |
- |
307 |
48 |
Drosophila |
55,000 |
- |
+ |
- |
170-300 |
23-58 |
Mammals |
61,300 |
+ |
+ |
+ |
56 |
19 |
Table 3: Comparative Data obtained from Luzzatto and Battistuzzi (1985).
As part of the kinetic investigation, the degree of coenzyme specificity was explored for G6PD in its ability to use NAD+ in lieu of NADP+. G6PD from Chlamydomonas demonstrated that NAD+ could only marginally be used as a coenzyme, if at all. The great majority of G6PD types can use both NADP+ and NAD+, albeit not indifferently (Luzzatto and Battistuzzi). However, various combinations of coenzyme specificities are seen in various groups of organisms. Preferential binding of NAD+ or NADP+ must obviously depend on differences within the coenzyme active site, suggesting these variations in turn have likely evolved in relation to different physiological roles of the enzyme. In more primitive organisms, for example Pseudomonas, this is especially clear where a NAD+-preferring G6PD is part of the catabolic Entner-Doudoroff pathway, whereas a NADP+-preferring G6PD supplies NADPH for biosynthetic processes (Luzzatto and Battistuzzi). In Leuconostoc mesenteroides NAD+ and NADP+ are associated with different conformations of the enzyme and the same is true for the rat mammary gland G6PD (Luzzatto and Battistuzzi). By contrast, G6PD from Bacillus cereus, Vibrio alginolyticus, yeasts, and fungi are uniformly NADP+- specific (Luzzatto and Battistuzzi).
Temperature and pH Optima
Similar to other NADP+-dependent dehydrogenases purified from C. reinhardii, G6PD exerts tremendous thermal stability, considering this primitive plant is indigenous to aquatic habitats with relatively mild climates. In vitro test results indicate G6PD exhibits maximum activity at 60 %C, but rapidly denatures beyond this thermal threshold.
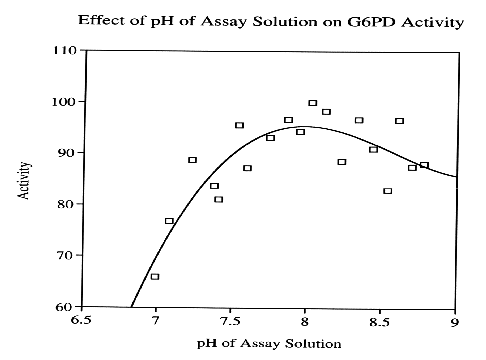
Traditionally, NADP+-dependent dehydrogenases are stable throughout a broad pH range. For most NADP+-dependent dehydrogenases, maximum enzyme activity was observed between the pH of 7.8-8.3 (Adams, et al., 1995). Isocitrate dehydrogenase from C. reinhardii demonstrates maximum activity around 7.4-8.1 (Beiler, et al., 1996).
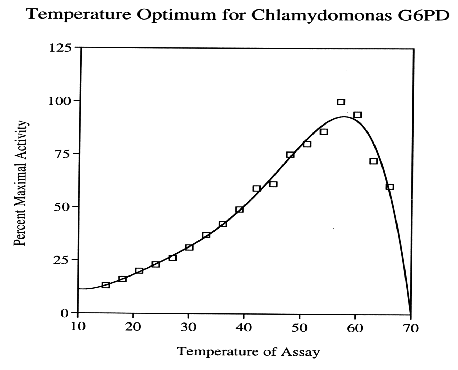
A potential explanation for the prevalence of pH tolerance in this series of enzymes may stem from the extensive conservation of G6PD and other NADP+-dependent dehydrogenases throughout all contemporary organisms, which include an incredibly diverse assortment of environments. Over time, G6PD has acquired structural resilience, a property that is the result of the enzyme being forced to adapt to a wide range organismal environments, containing a spectrum of many different temperatures and pH.
Divalent Metal Requirements
NADP+-dependent Isocitrate dehydrogenase and malic enzyme have demonstrated an absolute requirement for Mg2+ and Mn2+ from various sources (Maloney adn Dennis, 1977; Drincovich, et al. 1990) . In plants, it was suggested that divalent ions may play a key role in the regulation of the C4 pathway, not only at the decarboxylating step but also at the carboxylating step (Drincovich, 1990). On the other hand, heavy metals such as Cu2+, Zn2+, Hg2+, Pb2+ and Cd2+ have been proven inhibitors to NADP+-dependent dehydrogenase activity (Adams, et al., 1995). For G6PD, these divalent metals were surveyed to identify properties of the metal ions which may govern their interaction with the enzyme. No absolute divalent metal requirement was found for G6PD, although enzymatic activity was found to increase in the presence of Mg2+, Mn2+, Sr2+ and Ca2+. The observed increase in activity may not be a conclusiive enhancement and should be further investigated to verify the extent of the effects activating divalent metals have on G6PD activity. However, Cd2+, Hg2+ , Zn2+ and Ni2+ are toxic to G6PD activity. These particular heavy metals possibly deactivate the active sites of the enzyme, thus causing a complete loss of enzymatic activity. Furthermore, G6PD was neither negated nor able to utilize Cu2+ ions as electron acceptors, as indicated by the marginal effects observed in enzyme activity.
Use of Divalent Metal Ions by G6PD
| Ion* | Activity (%) |
| None | 100 |
| EDTA* | 102 |
| Manganese | 118 |
| Magnesium | 116 |
| Cadmium | 0 |
| Calcium | 123 |
| Cupric | 85 |
| Nickel | 0 |
| Mercury | 0 |
| Strontium | 115 |
| Zinc | 0 |
* 5 mM in assay solution
Molecular Size
G6PD has been characterized from a variety of different sources and consistently exists as an oligomer, with a predominance for dimeric and tetrameric structures. The molecular weight for the average G6PD monomer subunit is between 54 and 62 kDa, with more primitive organisms, exhibiting smaller monomeric subunits and more complex organism, such as mammals possessing larger monomeric subunits (Luzzatto and Battistuzzi).
Using a 6% polyacrylamide gel, the molecular weight for G6PD from Chlamydomonas was found to be approximately 180 kD. Extrapolating the previous values given for the G6PD monomer from other organisms, we surmise G6PD may be a trimer. However, no other organism has been found to contain trimeric G6PD, implying that the experimental value represents a tetramer composed of a relatively small monomeric subunit or a dimer with an extremely large monomeric subunit. Comparative subunit values reasonably project G6PD from the primitive Chlamydomonas to be a tetramer composed of a small subunit, which maintains the precedent of more primitive organisms having smaller G6PD monomeric subunits.
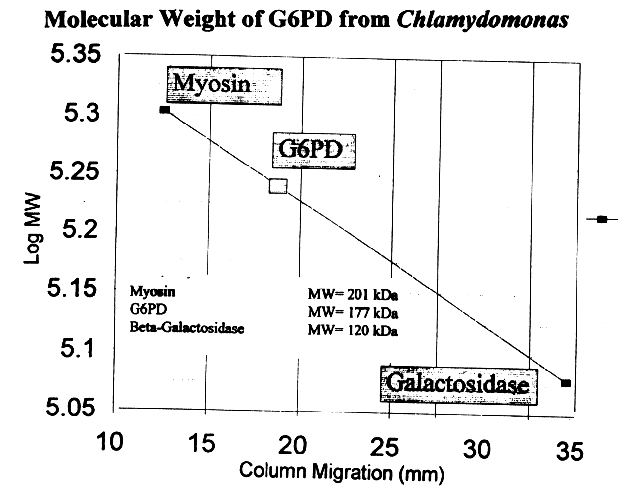
Figure 10: Molecular weight determination obtained
with PAGE, using the log MW corresponding to lane migration (mm).
Native G6PD Species
As was indicated earlier, some organisms have more than one functional form of G6PD. Using gel electrophoresis, a native activity stain revealed purified G6PD from C. reinhardtii expresses only one band, which means only one species of G6PD was observed. Thus, we conclude that we have purified G6PD from C. reinhardtii to apparent homogeneity, and learned functional G6PD from this organism may solely exist as a tetramer.

Figure 13: The presence of one band indicates Chlamydomonas reinhardii contains only one functional G6PD enzyme.
CONCLUSIONS
Experimental parameters were designed that quantitatively purified G6PD from Chlamydomonas reinhardtii using Amicon Matrex Blue A. Once purified, G6PD was characterized to explore its inherent enzymatic properties. G6PD from C. reinhardtii exhibited a broad pH optima, ranging from 7.6 to 8.3. Similarly, G6PD from C. reinhardtii is not heat labile, demonstrating maximum enzymatic activity at 60 %C. The kinetic survey for G6PD found the Km values for both substrates, G6P and NADP+, to be within the realm of published values for various organisms. The Km values for DL-G6P and NADP+ were 103.0_10.5 x 10-6 M and 4.8_0.153 x 10-6 M, respectively. G6PD from C. reinhardtii does not require a divalent metal ion to sustain enzymatic activity. Preliminary results suggest G6PD activity is enhanced in the presence of Mg2+, Mn2+, and Ca2+. However, G6PD cannot utilize Cu2+, Sr2+, or Zn2+ ions as electron acceptors. Moreover, Cd2+, Hg2+, and Ni2+ are toxic to G6PD activity. Electrophoretic analysis suggests only one functional species of G6PD exists for C. reinhardtiii, with an approximate molecular size of 180 kD, indicating the single form of the enzyme is most likely tetrameric.
Future investigations are necessary to confirm the data obtained in this series of experiments. With the recent elucidation of the three-dimensional structure of G6PD from humans, as well as Leuconostoc mesenteroides, a more extensive understanding of active site organization will contribute to the inter-species comparison and understanding regarding G6PD (Gomez-Gallego, 1996; Rowland, 1994). NADP+- dependent G6PD has proven its role as a crucial metabolic enzyme and serves scientific curiosity well as the enzyme becomes increasingly better understood, thus acting as a model enzyme for comparative research exploring other biochemical catalysts.
Acknowledgements
The researcher would like to express appreciation for Dr. John H. Williamson for his timeless patience, encouragement and committment to undergraduate research. The researcher would also like to thank Dr. A. Malcolm Campbell, Dr Elizabeth Harris, Dr. Pat Peroni, Dr. David C. Grant, Dr. Victoria McGovern, Dr. Donald Kimmel, and the Davidson College Department of Biology for their advice and suggestions throughout the course of this research. The researcher wishes to thank Renn Upchurch for her dedication and support. This research was supported by a grant to the Department of Biology from the Undergraduate Science Research Program of the Merck Company Foundation.
REFERENCES
Adams, A. B., P. H. Ewing, W. E.
Gordon, L. A. Patterson and C. J. Vargo. (1995) Characterization of NADP-dependent
enzymes from Chlamydomonas reinhardtii. Manuscript.
Amicon. (1989) Operating Instructions: Dyematrex Screening Kit for Protein Purification. Amicon Division,W.R. Grace & Company. - Conn. 24 Cherry Hill Drive, MA 01923.
Amicon. (1993) Dye-Ligand Chromatography. Applications-Method-Theory of Matrix Gel Media. Amicon Division, W.R. Grace & Company. - Conn. 24 Cherry Hill Drive, MA 01923
Arese P, and Flora A. (1990) Pathophysiology of Hemolysis in G6PD deficiency. Seminars in Hematology: 27: 1-30.
Beiler, R., Dozier, L., Edwards, B., Worley, S., and John H. Williamson. (1996) Characterization of NADP+-dependent Isocitrate Dehydrogenase from Brassica oleracea. Manuscript.
Campbell, N. A. (1993) Biology. (Third Edition). Redwood City: Benjamin/Cummings Publishing Company.
Crans, Debbie and Susan Schelble. (1990) Vanadate Dimer and Tetramer Both Inhibit G6PD from Leuconostoc mesenteroides. Biochemistry. 29: 6698-6706.
Drincovich, Maria Fabiana., Iglesias, Alberto A, and Carlos S. Andreo. (1991) Interaction of divalent metal ions with the NADP+-malic enzyme from maize leaves. Physiologia Plantarum. 81: 462-466.
Farr, Tracy J., Huppe, Heather C, and David H. Turpin. (1994) Coordination of Chloroplastic Metabolism in N-Limited Chlamydomonas reinhardti by Redox Modulation. Plant Physiol. 105: 1037-1042.
Fickensher, K., and R. Schiebe. (1986) Purification and Properties of the Cytoplasmic Glucose-6-Phosphate Dehydrogenase from Pea Leaves. Archives of Biochemistry and Biophysics: 247. 393-402.
Geer, B. W., D. Krochko, M.J. Oliver, V. K. Walker and J. H. Williamson. (1980) A comparative study of the NADP-Malic enzymes from Drosophila and chick liver. Comp. Biochem. Physiol. 65B: 25 - 34.
Gomez-Gallego, Felix., Pertierra, A.G., Mason, P.J., and Jose M. Bautista. (1996) Unproductive Folding of the Human G6PD-deficient variant A-. FAESB Journal. 10: 153-158.
Gordon, E., and C. Newman. (1995) Purification and characterization of isocitrate deydrogenase from Chlamydomonas reinhardtii.
Harris, E. H. (1989) The Chlamydomonas Sourcebook. 1st Edition. New York: Academic Press Inc.
Harris, E.H. (1997) Electronic Mail correspondence with Dr. Elizabeth Harris obtained March 28, 1997 from chlamy@acpub.duke.edu
Huppe, Heather C, and David H. Turpin. (1996) Appearance of Novel Glucose-6-Phosphate Dehydrogenase Isoforms in Chlamydomonas Reinhardtii during Growth on Nitrate. Plant Physiol. 110: 1431-1433.
Jeffrey, Jonathon., Persson, Bengt., Wood, Irene., Bergman, Tomas., Jeffrey, Rachel, and Hans Jornvall. (1993) Glucose-6-Phosphate dehydrogenase: Structure-function relationships and the Pichia jadinii enzyme structure. European Journal of Biochemisty. 212: 41-49.
Klein, Uwe. (1984) Localization of Enzymes in Chloroplasts from Chlamydomonas reinhardii: Enzymes of Glycolysis, the oxidative pentose phosphate pathway, and the citric acid cycle. Advances in Photosynthesis Research. III.6: 465-468.
Lowry, O.H., N.J. Rosebrough, A.L. Farr, and R.J. Randall. (1951) Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 193: 265-275.
Luzzatto L, Battistuzzi G. (1985) Glucose-6-Phosphate Dehydrogenase. Advanced Human Genetics. 14: 217-329.
Maloney, Robert J., and David T. Dennis. (1977) The Role of Divalent Cations in the Activation of the NADP+-specific Isocitrate Dehydrogenase from Pisium sativum L. Can. J. Biochem. 55: 928-934.
Rowland P, Basak A, Gover S, Levy R, and M. Adams. (1994) The three-dimensional structure of glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides refined at 2.0 A resolution. Structure, 1073-1087.
Stryer, Lupert. (1995) Biochemistry. New York: W.H. Freeman and Company.
Takizawa T, Huang IY, Ikuta T, Yoshida A. (1986) Human glucose-6-phosphate dehydrogenase: Primary structure and cDNA cloning. Proceedings of the National Academy of Science: 83: 4157-4161.
Vulliamy T, Mason P, Luzzatto L. (1992) The Molecular Basis of Glucose-6-Phosphate Dehydrogenase deficiency. Trends in Genetics: 8: 138-143.
Williamson, J. H., D. Krochko and M. M. Bentley. (1980a) Properties of Drosophila NADP-isocitrate dehydrogenase purified on Procion Brilliant Blue Sepharose 4B. Comp. Biochem. Physiol. 65b: 339-343.
Williamson, J. H., D. Krochko and B. W. Geer. (1980b) 6-Phosphogluconate dehydrogenase from Drosophila melanogaster: I. Purification and properties of the A-isozyme. Biochem. Genetics 18: 87-101.
Williamson, J.H. (1986) G6PD of Drosophila melonogaster. Glucose-6-Phosphate Dehydrogenase. New York: Academic Press Inc.
![]()
![]()
© Copyright 2000 Department of Biology, Davidson
College, Davidson, NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu