Routine chemicals were obtained from Sigma Chemical Company. Ammonium
sulfate was obtained from Fisher Scientific
Company, pre-stained SDS-PAGE standards and pre-cast gels were obtained
from BioRad, and Matrex Red A Gel (Figure 3) was
obtained from Millipore Corporation. The Oregon R strain of Drosophila
melanogaster (homozygous for IdhF; Fox 1971) was
cultured on synthetic medium (Carolina Biological Supply) at 25°C,
collected 0-4 days after eclosion and stored at -20°C until used.
Figure 3: Partial chemical structure of Matrex Red A Gel (Dye-Ligand
Chromatography: Applications - Method - Theory of Matrex Gel Media.
Amicon, Inc. Beverly, Mass. 1993.)
Isocitrate dehydrogenase was purified using a two-step procedure derived
from Ni, et al., (1987) which included ammonium sulfate
fractionation and Matrex Red A affinity chromatography. IDH was eluted
by washing the Matrex Red A column with a gradient of
NADP+ and isocitrate. The column was packed by first diluting
the Matrex Red A slurry with affinity chromatography buffer (10 mM
K-phosphate, pH 7.0; Ni, et al., 1987) and then pouring this
mixture into a 1.6 X 21 cm glass column until the matrex beads were
compact. The column was placed in a 4°C chromatography cabinet
and washed with 50 mL of 1 M NaCl, 50 mL of 4 M urea, and,
finally, with 100 mL of affinity chromatography buffer.
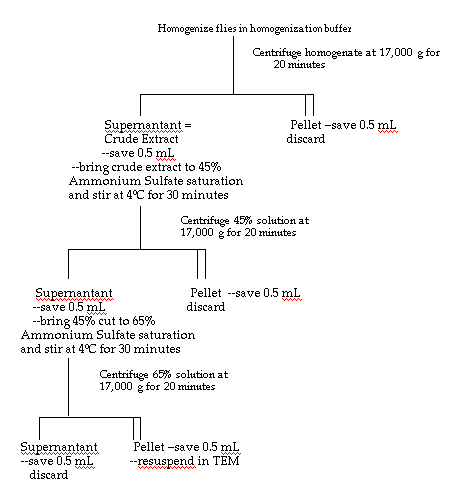
Enzyme Purification
Thirteen and a half grams of the Oregon R strain of Drosophila melanogaster
were homogenized in 45 mL (approximately three
volumes) of homogenization buffer (25 mM Hepes - NaOH, pH 7.6; Ni,
et al., 1987) and centrifuged at 17,000 g for twenty minutes.
The pellet was discarded and the supernatant was measured and labeled
"crude homogenate." The crude homogenate was brought
to 45% saturation with ammonium sulfate and stirred for thirty minutes.
After centrifugation of the 45% preparation at 17,000 g for
twenty minutes, the supernatant was measured and brought to 65% ammonium
sulfate saturation while stirring for thirty minutes.
The 65% solution was then centrifuged for twenty minutes at 17,000
g. The pellet resulting from this final centrifugation was saved and
resuspended in 5 mL of TEM buffer (0.1 M Tris, 5 mM EDTA, 1 mM mercaptoethanol,
pH 7.6). A small volume (0.5 mL) of each
preparation was reserved for enzyme activity and protein concentration
assays.
The resuspended 65% pellet was further diluted five-fold with TEM buffer
and slowly pumped onto the column. The column was
washed overnight with affinity chromatography buffer (153 mL) to remove
unbound materials. IDH was eluted by washing the
column with a 150 mL gradient of NADP+ and isocitrate in
affinity chromatography buffer (0-1 mM NADP+ and 0-24 mM isocitrate).
The column was recharged by washing with 50 mL of 4 M urea and 100
mL affinity chromatography buffer. All procedures were
carried out at 4°C. Figure 4 is a complete diagram of this purification
scheme.
- Dilute the resuspended 65% pellet with TEM and pump over Matrex Red A Column.
- Pump 50 mL of Affinity Chromatography Buffer (ACB) over column.
- Pump 100 mL of a NADP+/Isocitrate gradient over column.
- Pump another 50 mL of ACB over column.
Protein Determination
Protein concentrations were determined by the method of Lowery, et
al., (1951). Triplicate assays were performed for each sample and bovine
serum albumin was used as the protein standard.
Enzyme Activity Assays
IDH activity was determined by monitoring the reduction of NADP+
at 340 nm in 1 mL assays containing 0.1 M Tris, 0.144 mM NADP+,
1
mM MgCl2, and 0.23 mM DL-isocitrate with a Beckman Model 35 spectrophotometer
equipped with a heated cuvette chamber and water
circulator set at 30°C. Routinely, 10 µL samples of substrate
were used to initiate the reactions. Activity (initial velocity) was calculated
as
micromoles of NADP+ reduced per minute. Specific activities were determined
as micromoles of NADP+ reduced per minute per microgram of
protein.
Sample Concentration
Fractions with peak IDH activity were concentrated using Eppendorf Centrifugal
Filter tubes (Brinkman Instruments, # 022-65-042-4) with 30 kDa
exclusion filters. The material to be concentrated was placed above
the filter and centrifuged at 3,000 g for 60-90 minutes. The resulting
concentrate (250 - 500 µL) was recovered and used for activity
and protein assays and gel electrophoresis.
Polyacrylamide Gel Electrophoresis
The native size of IDH was determined using non-denaturing polyacrylamide
gel electrophoresis. Gels (either 7.5% or 10%) were loaded
symmetrically, run at 200 volts for about 45 minutes at 4°C, and
cut in half. One half was stained for IDH activity, ibidem, using
0.06 M Tris
buffer, 0.35 mM NADP+, 2.4 mM nitro blue tetrazolium, 0.6
mM phenazine ethosulfate, and 0.39 mM of DL-isocitrate, at 30°C in
the dark
(Fox, 1971). Protein bands were detected by staining the other half
of the gel with Coomassie Brilliant Blue R at room temperature for thirty
minutes and de-stained in a 5% methanol - 7.5% acetic acid solution
for one to two days.
Subunit sizes of IDH were determined by SDS-PAGE. Samples of concentrated
IDH were heated to 95°C, loaded on a 10% gel, and submitted to
200 volts for 45 minutes at room temperature. Gels were stained with
Coomassie Brilliant Blue R for 30-60 minutes and destained for one to two
days.
Substrate Kinetics
Km values were determined by measuring the reduction of NADP+ at
340 nm and 30°C at seven concentrations of substrate or coenzyme while
holding all other assay conditions constant. Km values were
determined by Lineweaver-Burke analysis.
Divalent Metal Ions
Assay solution was prepared with deionized water and ultra-pure buffer
ingredients (Sigma) to determine whether IDH has an absolute requirement
for divalent metal ions.