A. Cloning of the pmh1Gene
I. Subcloning of the pmh1gene
Before the pmh1 gene could be reconstructed into one continuous piece of DNA, six lambda genomic DNA samples (prepared by Stephanie Moses, í97) containing the putative pmh1 gene were analyzed for their cloning suitability (Coble, 1998). After choosing the best sample, subcloning was carried out on the putative pmh1 gene. A 8.4 kb Sal I fragment (SF) was isolated and cloned by Allison Coble (í98) (Figure 6) and a 6.4 kb BamH I fragment (BF) was cloned by Dr. Virginia Armbrust (Figure 7). These two fragments have an overlapping Sal I-BamH I region which is 0.8 kb. Lindsey Cohen (í99) then generated several smaller subclones in pSK-(From Stratagene) and pZero (from Invitrogen). A total of six subclones were generated, of which half contained inserts of various length derived from BF while the other half contained SF derived inserts. The main objective of generating these smaller subclones was to facilitate the creation of the final constructs since smaller sized fragments are much easier to manipulate and sequence.
II. Analysis of the pmh1 gene prior to formation of final constructs
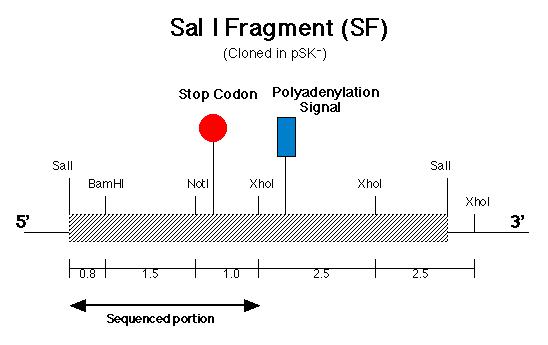
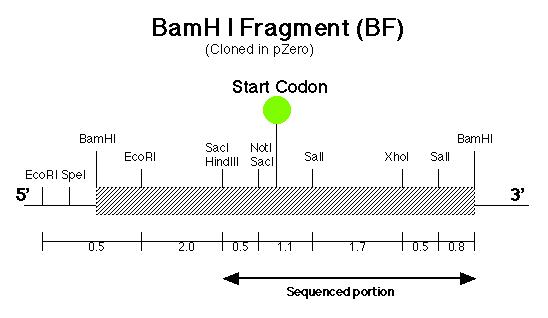
The pArg7.8 insertion in the pmh1 gene prevented the 4.4 kb message from being detected by probe E (Figure 5). To rescue the phenotype through complementation, the pmh1 gene that codes for the 4.4 kb message must be reconstituted and transformed into the pmh1 mutant. Analysis of the sequenced putative pmh1 genomic DNA and pmh1 cDNA revealed that the largest open reading frame is 3.0 kb. Since the pArg7.8 insertion site is found within this open reading frame, the mutation is localized within this coding region. The start codon on this open reading frame is located on the BamH I fragment (BF) cloned by Dr. Armbrust (Figure 7) while the stop codon is located on the Sal I fragment (SF) cloned by Allison Coble (Figure 6). In addition, it was found that the polyadenylation signal is located 1.0 kb downstream of the stop codon. This suggests that the 1.0 kb fragment between the stop codon and the polyadenylation signal is the 3í-untranslated region (Figure 6).
Since the start and stop codons of the
open reading frame are located on the BF and SF subclones respectively,
relevant portions of SF and BF that contain the open reading frame must
be cut and spliced together in order to make final constructs of the putative
pmh1 gene. Besides cloning the open reading frame, it is also crucial
that the upstream promoter sequence and transcription initiation sites
be included in the clones since these sequences facilitate transcription.
In addition, the polyadenylation signal has to be present within the clone
so that a poly-A-tail can be added to make a complete transcript.
III. Formation of final constructs
III-1) PCR of pmh1 SF
The first part of my project in the building
of construct A involved cloning the fragment derived from SF which contains
the stop codon and the polyadenylation signal. To accomplish that, Polymerase
Chain Reaction (PCR) was carried out to amplify that fragment. Since the
location of the polyadenylation signal is known, PCR primers were made
to flank the sequence immediately downstream of the polyadenylation signal
and also at the 5í-end sequence immediately upstream of the BamH I restriction
site (Figure 6). The 3í-end primer contained an additional 10 nucleotides
which coded for a Kpn I restriction site. The Kpn I site is needed to generate
sticky ends on the amplified DNA for cloning purposes. If PCR using
SF as the template were successful, we would obtain an amplified 2.8 kb
DNA fragment spanning the 5í BamH I restriction site to the 3í Kpn I restriction
site (due to the modified primer).
Before the BamH I-Kpn I PCR product could be
amplified, the template DNA was obtained. The template DNA came from SF
cloned in the pSK- plasmid (pSK-/Sal I). Thus, cell
cultures were grown and alkaline lysis mini-preps were conducted on pSK-/Sal
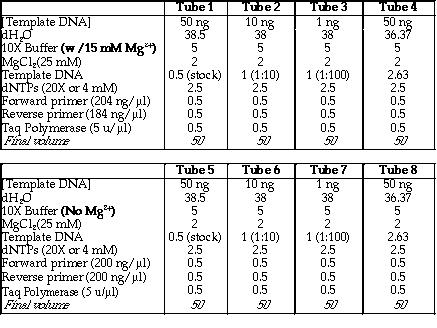
I. Prior to running the PCR, I had to determine the the optimal amount
of pSK-/Sal I template DNA and Mg2+ needed
in the reaction. The amount of Mg2+ used in PCR is crucial in
obtaining an optimum yield. Thus, two sets of experiments were carried
out. Figure 8 is a table that shows the volume of each reagent used in
both sets of PCR experiments.

In order to ensure that the results were reproducible,
PCR was repeated using the exact conditions from tube 3. All the reagents
and volumes used in tube 3 remained constant. Five separate PCR reactions
were completed and the contents from all the tubes were pooled. Another
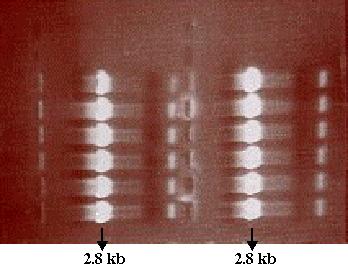
0.7% agarose gel loaded with the PCR tube 3 product was run to verify the
presence of amplified DNA. Analysis of the gel showed that PCR was successful
and the desired PCR product using SF as template was amplified (Figure
10). Now that the presence of the desired PCR product had been verified,
the PCR product was purified via electroelution (Figure 11).

III-2) Cloning of PCR product in pSK-
The next step in my project involved cloning
the PCR product in pSK-. Since the PCR product is flanked by
BamH I and Kpn I restriction sites, restriction digestions using both enzymes
were performed. Previous experiments had shown that a double digestion
could not be performed simultaneously because BamH I required a buffer
with a high salt content (100 mM NaCl) while Kpn I required a buffer with
low salt content (50 mM KCl). Thus, two separate digestions were conducted.
Since PCR product would be cloned in pSK-, the pSK-
plasmid was also digested with both restriction enzymes. The PCR product
and pSK- were first digested with the low salt cutter Kpn I,
"cleaned" , and then digested with the high salt cutter BamH I and then
"cleaned" again. The DNA samples were digested in three duplicate sets
before being pooled for "cleaning". Quantification of the digested
and cleaned DNA samples was carried out on a spectrophotometer and the
absorbance and concentration for each sample were determined.
After generating sticky ends on both the vector
(pSK-) and insert (PCR product), two ligation reactions were
performed. The first reaction had a 2:1 insert to vector ratio while the
second set contained a 4:1 insert to vector ratio. After transformations,
five colonies were detected on the plate with the 4:1 insert to vector
ratio ligation and one colony growing on the 2:1 insert to vector ratio
ligation. These colonies were picked and grown in LB-Amp media and alkaline
lysis mini-preps were carried out to isolate the plasmids from the cells.
To verify the presence of the PCR product in the pSK- vector,
a Not I digestion was carried out. The digestion was run on a 0.7% agarose
gel electrophoresis to verify the PCR product. Results obtained from the
first gel, hinted that cells from tube 1 and tube 6 were successfully transformed.
However, due to overloading of the samples, as well as the poor resolution
of the 1 kb ladder, the data was inconclusive (data not shown).
Another 0.7% agarose gel electrophoresis with just plasmid samples from
tube 1 and tube 6 was run. Analysis of the gel revealed a 1.6 kb fragment
and a 4.0 kb fragment (Figure 12). These two fragments proved that the
ligation reaction had been successful. Thus, the PCR product was confirmed
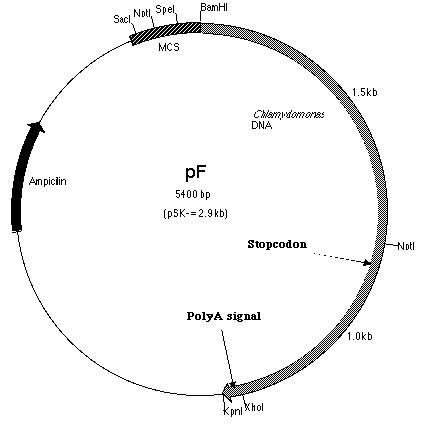
cloned within the pSK- plasmid. This newly cloned plasmid was
labeled pF (Figure 13).

III-3) Construction of pmh1 Construct A
With the PCR product successfully amplified
and cloned (pF), construct A could now be made. Construct A is a fusion
of the PCR product with pE. pE (Figure 14) is one of the six subclones
constructed by Lindsey Cohen (í99) and it contains a truncated BF insert
cloned within the pSK- plasmid. The start codon is located within
this truncated insert. The PCR product on the other hand, is derived
from SF. Thus, construct A (Figure 15) contains the putative pmh1
gene that includes the start and stop codons as well as the polyadenylation
signal. It was hoped that the additional 0.1 kb fragment upstream of the
start codon contained the promoter sequence.
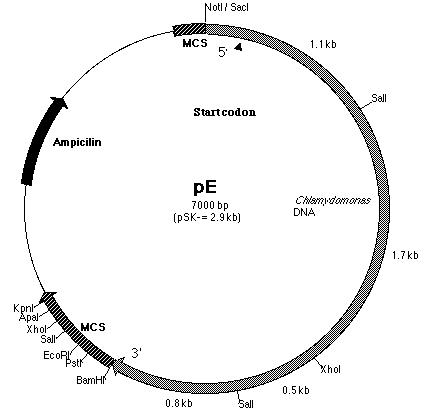
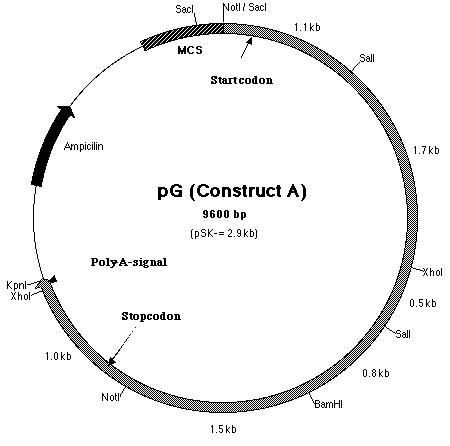
Figure 15: Restriction map of construct A which is also known as pG.
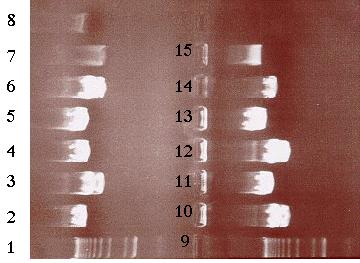
Building construct A requires ligating the PCR product into pE. Both pE and the PCR product were digested with BamHI and Kpn I restriction enzymes to generate sticky ends. Next, a ligation reaction at a 8:1 insert to vector ratio was conducted to insert the PCR product into the 3í-end of the 3.8 kb insert in pE (Figure 14). Heat shock transformation of the ligation was then carried out and nine colonies were observed after overnight incubation. A Not I digestion was done on each of the nine plasmid mini-preps for the transformed colonies to verify the presence of the insert in pE. A 0.7% agarose gel electrophoresis loaded with the restriction digestions was ran to identify the plasmids. Analysis of the gel showed that plasmid samples 2, 5, 6 and 8 (lanes 3, 6, 10 and 12 on the gel) contained a 6.0 kb and 4.0 kb fragment. Both these fragments indicated that the ligation reaction was successful (Figure 16). The PCR product had been cloned into pE and construct A was ready. The newly formed construct A was labeled as pG (Figure 15).
III-4) Construction of pmh1 Construct B
Work was also done to build construct B (Figure
17). Construct B is construct A with an additional 2.5 kb fragment ligated
to the 5í-end of the insert. If construct A failed to work due to the absence
of the promoter region, it is believed that the additional 2.5 kb fragment
which lies upstream of the start codon should contain the promoter sequence
and possibly the enhancer region as well. In order to create construct
B, plasmid pD (Figure 18) was used. Plasmid pD is a pZero plasmid that
contains a 6.3 kb insert. This insert, like pE, is derived from BF. However,
it contains the additional 2.5 kb Not I-Eco RI fragment upstream of the
pE insert. To build construct B, Kpn I and BamH I restriction enzymes were
used to generate sticky ends from both pD and the PCR product. Both the
insert (PCR product) and the plasmid vector (pD) were ligated together
to produce construct B. Although the PCR product had already been cloned
in pF, pF was not used because if the PCR product was digested with BamH
I and Kpn I, two fragments of almost equal sizes would be generated. The
digestion would generate 2.8-2.9 kb fragments derived from the PCR product
and the pSK- plasmid vector (Figure 13). Hence, the PCR product
could not be resolved from the pSK- plasmid vector in an agarose
gel electrophoresis.
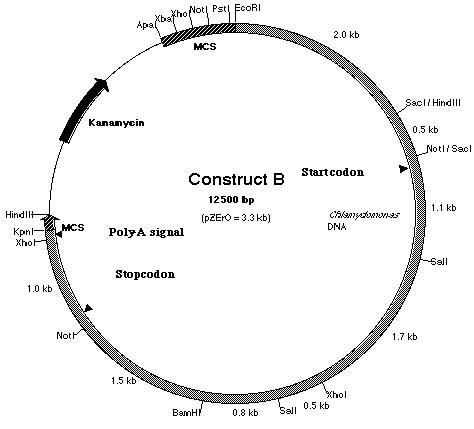
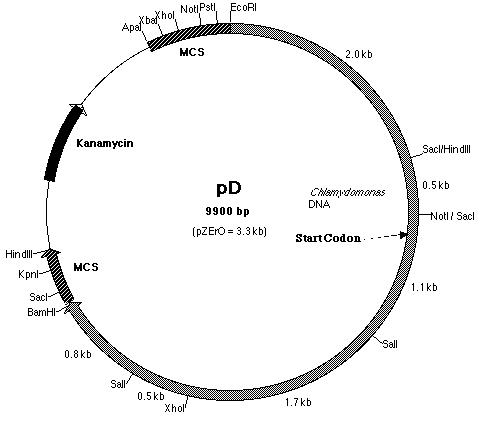
Figure 18: Restriction map of pD plasmid which contained
the 5' half of the pmh1 gene. The start codon is located on pD.
Many complications surfaced in the process of cloning construct B. First, contamination in the T4 DNA ligase led to the growth of foreign colonies on the transformation plates. On a casual glance, the morphology of the colonies resembled the expected transformed E. coli colonies. However, careful observation revealed that the foreign colonies grew slower and also appeared to be more opaque than E. Coli colonies. In addition, no plasmid DNA was detected in the gel electrophoresis of the plasmid mini-preps (data not shown). Since contamination of the T4 DNA ligase is extremely rare, we spent several weeks trying to determine the source of the contamination.
Once the contamination problems were eliminated,
several transformations of construct B were conducted using various competent
cells. The first few transformations were done using JM109 competent
cells from Stratagene. We then switched over to MAX efficiency STBL2 competent
cells from GibcoBRL. Finally, we decided to use an ultracompetent cell
, Epicurian Coli XL10-Gold from Stratagene. The results obtained from using
all three competent cells were identical and the transformation failed
to produce the desired clone. In all three transformations, a single smeared
band of 6.0 kb was detected on the gel (Figure 19). This single band is
smaller than pD (9.9 kb). It is possible that construct B is highly unstable
and that recombination might have occurred after transformation. In addition,
the insert in construct B is three times the size of the plasmid and the
total construct is around 13.0 kb in size. Thus, the size of constuct B
could have reduced the cloning efficiency and probably resulted in other
complications.
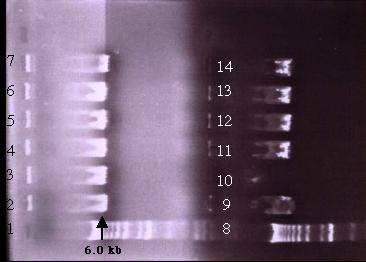
Figure 19: A 0.5% agarose gel electrophoresis of the Xho I digested plasmid mini-prep samples. On both half of the gels, lane 1 and 8 is the 1 kb ladder while lanes 2-7 and 9-14 are the plasmid samples. The bands indicated the presence of a DNA fragment roughly 6.0 kb in size in each lane. This is smaller than pD. If transformation was successful, three bands of 5.1 kb, 4.0 kb and 3.3 kb would be detected. Failure of transformation could be due to the size of the construct.
During transformation, autolysin was added to help break down cell walls so that the targeted vector can enter the cell and transformation can occur. Autolysin was not used for the first several transformation attempts. This was due to observations under the light microscope that suggested pmh1 lacked cell walls (Russo, personal communication). Although several variables such as polyethlyene glycol concentrations, nit1 concentrations, duration of vortexing cells with glass beads and the amount of beads used were altered, no colonies grew on the NH4+ free plate. This indicated failure of transformation.
Autolysin was then prepared and used in subsequent
transformations. Once again, no colonies were observed. Failure of transformation
could be due to the excessive heat generated by the construction work in
the Dana science building. Another transformation was then carried
out with autolysin and the plates were incubated in a 22 degrees Celcius
growth chamber. Transformation with strain A54 was also carried out simultaneously
to determine toxicity of the autolysin. Since strain A54 is capable of
surviving and growing in a nitrogen-free medium, it should grow on the
plates unless any of the reagents used in the transformation were toxic.
Results showed that the A54 grew well in the nitrogen-free media. In addition,
green colonies were spotted growing on the experimental plates. These
colonies were individually selected and streaked on NH4+
free plates using sterilized toothpicks. Roughly 150 colonies were selected
and transferred using this method. Once the colonies were allowed to grow
for a week, it was apparent that they were contaminated with a foreign
organism (Figure 20). The foreign organism appeared to be bacterial (Wessner,
personal communication) and the Chlamydomonas cells seemed to grow
on the surface of the contaminant (Figure 21).


Figure 21: Chlamydomonas colonies that were streaked
out on a NH4+-free plate. These streaked patches
contained a mixture of Chlamydomonas cells and the bacterial contaminants.
A decision was made to try and isolate the
transformed colonies from the contaminants. Under the dissecting microscope,
individual cells in a colony were teased away from the contaminants and
they were streaked out on fresh NH4+-free plates.
After several attempts, it was concluded that the cells could not be separated
from the bacterial contaminants and the contaminants usually overwhelmed
the plate before the cells had a chance to grow (Figure 22).

Figure 22: The individual Chlamydomonas cell that was isolated from a colony and then plated on a fresh NH4+-free TAP plate are being overwhelmed by the bacterial colony. All that is seen here is a white lawn of bacterial contaminants.
In a separate experiment, some of the mixture of cells were streaked out on plates that were coated with amphicilin and kanamycin which killed the bacterial contaminants. Results indicated that at high antibiotic concentrations (100X), the bacterial contaminants were absent. However, no Chlamydomonas colonies grew on those plates either. This suggested the "transformed" cells were absorbing the NH4+ waste secreted by the bacteria. Thus, without the bacteria, the Chalamydomonas cells could not survive in the NH4+-free environment. To answer that question, an experiment was devised and performed to determine whether the Chlamydomonas cells were really transformed. The data showed that pmh1 alone could not survive on a nitrogen-free media. However, if the contaminants were mixed with pmh1, the cells grew well (Figure 23). This experiment proved that transformation never occurred and the colonies that were "transformed" were actually dependent on the contaminant for survival and growth.

Figure 23: Experimental plate to determine whether the
Chlamydomonas cells were transformed (23A). pmh1 cells were
unable to grow alone on the NH4+-free TAP (23B).
However, in the presence of the bacterial contaminants, abundant growth
was observed (23C). This indicated that the pmh1 cells were surviving
on a NH4+ source secreted by the bacterial contaminants.
C. pmh1 Sequence Analysis Via MacDNAsis
Since the pmh1 gene had been sequenced,
sequence analysis of the pmh1 genomic DNA, cDNA and amino acid sequence
was carried out via MacDNAsis. First, the genomic DNA was scanned for several
restriction enzyme cut sites. This analysis enabled us to verify the cut
sites obtained from the restriction mapping that was done prior to sequencing.
Results indicate that all the cut sites were correctly predicted via restriction
mapping. However, an additional Sal I site was found in between the BamH
I and Not I restriction site in SF (Figure 6). The presence of the Sal
I site is most probably due to a sequencing error. Using the pmh1
cDNA, we also identified the correct open reading frame for the pmh1
gene. The start codon was determined to several bases downstream of the
Not I/Sac I restriction site (Figure 7) while the stop codon was located
downstream of the Not I site in SF (Figure 6). The pmh1 amino acid
sequence was used to predict transmembrane domains via the Kyte and Doolittle
hydrophobicity plot. A total of ten hydrophobic transmembrane region as
well as a phosphorylation domain were detected as is typical of P-type
ATPases (Russo, personal communication; Figure 24).
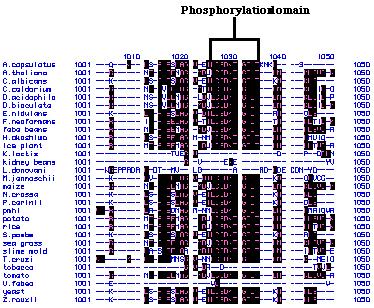
A Genbank search was then carried out to to
identify other proteins that displayed a strong sequence homology with
the pmh1 sequence. The protein sequences derived from twenty nine
other organisms were extracted and then a multiple sequence alignment was
carried out . This multiple sequence alignment not only identified all
the highly conserved regions in pmh1, it also allowed us to create
a phylogenetic tree based on the relational similarities of the sequences
being compared (Figure 25). Further analysis of the multiple sequence alignment
and the phylogenetic tree will be described in the discussion section of
this paper.
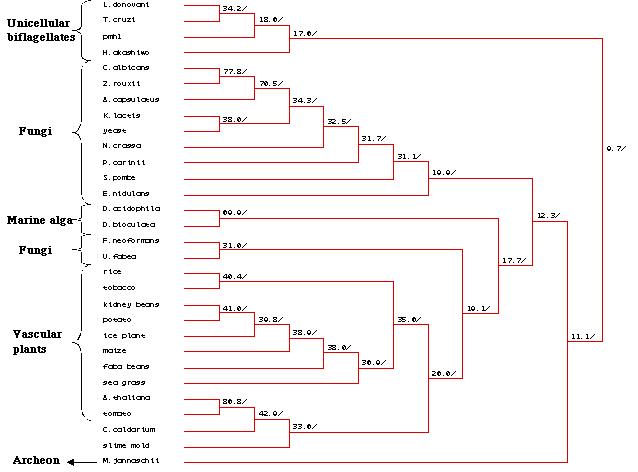
Figure 25: Phylogenetic tree created by MacDNAsis for
pmh1 and twenty nine other organisms using the Higgins and Sharp
model (Higgins et al., 1988).