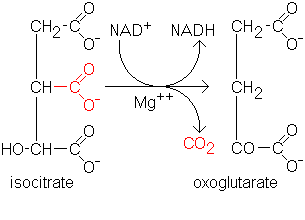
Figure 1. The conversion of isocitrate to alpha-ketoglutarate by IDH.
In the last five years, there has been a tremendous flow of information from sequencing centers into the sequence databases. As a result, a substantial portion of bioinformaticists is involved in writing programs to facilitate the collection and organization of the sequence data. With the completion of the Drosophila melanogaster genome, and that of the human genome not too far away, we can now concentrate on the next step -- the farming of the sequence data for research purposes.
The sequence databases have already been in use for certain purposes. Newly discovered genes and proteins are often assigned names and functions based on their homology with other proteins in the database. Gene-finding programs have been trained to look for coding seqences of DNA using the databases as input. Functional genomics studies have employed micro-array technology to assess the differential expression of genes during various cellular events (De Risi et al., 1997). As more and more sequences from a wide variety of organisms are entered in the database, we can envision yet another application -- tracing the evolution of proteins.
Our objective was to develop a system to investigate the evolution of a protein and its functional relationships with other proteins in the database. The system would involve the use of public domain sequence similarity programs, as well as a new program called Divide and BLAST, which was written as part of this thesis.
The National Center for Biotechnoogy Information (NCBI) has developed two programs that can be used to search for conserved domains -- PHI-BLAST (Pattern Hit Intiated BLAST - Zheng et al., 1998) and PSI-BLAST (Position-Specific Iterated BLAST - Altschul et al., 1997). Both programs are based on the BLAST (Basic Alignment and Search Tool) program (Altschul et al., 1990) and do several iterations of BLAST to fulfil their purpose. Their output is often useful, but they suffer from the same limitations as BLAST, which sacrifices thoroughness for speed. Remote relationships are hard to detect, and proteins that are closely related align completely, giving no idea of the location of the highly conserved areas. Divide-and-BLAST uses a divide-and-conquer approach to attempt to overcome some of the limitations of the BLAST family of programs (Karnik, 2000).
Our test proteins for this study were isocitrate dehydrogenase 1 (IDH1 - EC 1.1.1.42) and caspase 3(apopain). Isocitrate dehydrogenase 1 catalyses the conversion of isocitrate to alpha-ketoglutarate, with NADP acting as a cofactor (Hurley et al., 1989, Fig. 1). It is found in the cytoplasm and present in a wide variety of organisms, both eukaryotic and prokaryotic. It is known to be evolutionarily related (by sequence similarity) to isopropylmalate dehydrogenase (Imada et al., 1991) and tartrate dehydrogenase (Tipton and Beecher, 1994). We used IDH1 as our model protein, since we could predict our results to a great extent.
Our second test protein was caspase 3. Caspase 3 is part of the cysteine protease family of proteins, also called caspases, and is one of the key proteins involved in apoptosis (Cohen, 1997). Since apoptosis is only seen in eukaryotes, we hoped to find a prokaryotic precursor of caspase 3 that eventually evolved into the caspase 3 protein that is so vital in apoptosis.
Back to Table of Contents