Ordering Information:
The kit is the Total RNA Safekit
from Bio101
the 50 prep kit
cat# 1008-200
phone # is 1-800-424-6101
(www.bio101.com)
This is the Student Handout Written by Dr. Karen Bernd, at Davidson College. Email: kabernd@davidson.edu.
Reading: Molec Cell Biology pg 240-241 "Analyzing specific Nucleic Acids in Complex Mixtures" through to "specific RNAs…"
This week's lab looks incredibly long. That is mainly because each step is written out. Make sure to read over the protocols and have any technical (or philosophical) questions ready.
Isolation of total RNA from yeast
In order to further characterize the mating reaction you will be isolating total RNA from TWO cultures of DLY1. One culture contains cells in logarithmic growth phase grown in YEPD. The other contains 'log' cells, in YEPD, that have been treated for over 3 hours with a factor. (so what should they be doing?) Next week you will determine the effect of a factor on transcription of FUS1, a gene involved in the mating reaction, and IPD2, the gene encoding isocitrate dehydrogenase by performing dot blot analysis on the samples.
One of the biggest problems in most experiments is being able to tell the difference between a 'result' and an artifact of the experiment. Previously we used growth on different types of media as positive and negative controls to detect complementation and, therefore, mating. In these experiments we will be detecting an mRNA that is 'constitutively' expressed (always expressed) as a positive control to show that the methods are working. Our positive control is the IDP2 gene. As you prepare the RNA consider what you will use for a negative control, what it will show, why it is appropriate for this experiment.
RNA isolation is tricky. You must get rid of RNases
Your objective is to isolate a large quantity (perhaps 100 whole micrograms) of high molecular weight, undegraded total RNA. Read all instructions before even touching anything. Make sure you have cleaned your bench space before beginning.
The most important step in RNA isolation is to remove as many sources of RNases from your work area as possible. RNases are enzymes whose entire purpose is to degrade RNA. They are very stable proteins that can withstand high temperature and they are EVERYWHERE (yes, be paranoid!). ANY contamination with RNase will destroy your sample and pretty much wreck your afternoon, experimentally speaking. The samples you isolate today will be the basis for the blot you perform this week and next week AND will be saved so that you can use them as controls in your experiments that characterize you mutants later in the semester. Extra caution now will save you a bunch of work then.
A main source of RNase contamination is your hands. Since it is out of the question to bake your hands at 210° for 15 hours to inactivate the enzyme, you must do everything you can to eliminate contact between your skin, things your skin has touched, and your precious samples. Clear your bench of all but the bare essentials. Once you have broken open the cells the RNAs are released and are susceptible to degradation by RNAases. Wear gloves and wash down your entire area with 'RNase Erase' (special detergent that helps a little). All microfuge tubes and pipet tips have only been touched with gloves and have been sterilized extensively. The protocol is not difficult but you must be organized and careful.
Preparing yeast cells by making spheroplasts
Yeast contain a hard cell wall. Spheroplasts are yeast cells where the cell wall has been enzymatically degraded.
1) Each group receives 30mls of non-induced DLY1 cells and 30mls of a factor induce DLY1 cells (a factor was added to 1ug/ml of media approximately 3 hours before class) in 50ml conical tubes tubes. All cultures are in log phase growth.
2) Label the non induced DLY1 tube "N" and the induced cell tube "I" also add a distinguishing mark so you can identify your tube.
3) Place N and I tubes in centrifuge (balancing it). Centrifuge at 5000rpm for 5min to pellet the cells. There will be more than one group using the centrifuge, try to be good 'lab citizens'
4)While spinning, get other supplies
- yellow and blue tips for pipetmen (boxes)
- 2ml Microfuge tubes
- Potassium Phosphate/Sorbitol buffer
The next 3 steps are called a wash--they serve to move the cells from the growth media into a solution that is correctly buffered for the next procedure.
5) Remove tubes from centrifuge. Carefully pour supernatant into waste container. Try not to disturb pellet. pellet. Add 1ml of Potassium Phosphate/Sorbitol buffer. Resuspend the pellet by pipetting.
6) Label one 2ml microfuge tube 'N' and one 'I'. Transfer the cells from the conical tubes to the appropriate centrifuge tube.
7) Place the microfuge tubes in the microcentrifuge at your bench (balancing tubes). Spin at 80% for 3min.
8) Pour off supernatant (without disturbing pellet). Remove remaining supernatant with pipetman. Resuspend pellet in 1ml Potassium Phosphate/Sorbitol buffer.
9) Take tubes to hood. Add:
- 3µl b mercaptoethanol (a reducing agent that smells very bad)
- 320 µl of lyticase (enzyme that degrades cell wall components)
10) Place tubes in shaker at 30°C for 15min.
The cells are now spheroplasts. Without a cell wall they are alive but strucutrally much weaker. Care must be taken so that the cells don't burst before you want them to. (How could the buffer they are in help keep them intact?)
NOTE: yeast will repair their cell wall if the enzyme is removed so we cannot just keep a stock of 'wall free' yeast.
You must now get the cells ready for the next set of steps by washing away the lyticase and b-MeOH.
11) Spin in microcentrifuge at 40%. Remove supernatant by pipetting (discard in hood to contain the smell).
12) Add 500µl potassium phosphate/sorbitol buffer. Repeat step 11 (one time).
13) While centrifuging get ice bucket from side bench containing:
2 microfuge tubes containing glass beads1 tube of Solution 1 (1.6ml)1 tube of Solution 2 (400µl)1 tube isopropanol (1.6ml)1 tube of Solution 3 (400µl)1 tube of sterile dH2O (1.6ml)
14) Clean RNA isolation area with 'RNAse away' solution (1 spray bottle for lab)
Isolating RNA
| FROM THIS POINT ONDO NOT TOUCH ANYTHING ON YOUR BENCH WITHOUT WEARING GLOVES.DO NOT 'DOUBLETOUCH' TIPSDO NOT WALK AROUND WITH OPEN TUBESDO NOT LAUGH, TALK, OR BREATHE OVER OPEN TUBES
(To quote the movies "be afraid--be veryafraid")
|
15) Resuspend pellets from step 12 in 800ul Solution 1 by gently flicking tube.
16) Pour glass beads from tube in icebucket into tube containing cells
17) Add 200µl of Solution 2 to each cell+bead tube. Solution 1 and 2 contain buffers and chaotropes that will help stabilize the RNA in solution).
18) Using the 'Turbo vortexer" (side bench) vortex the cells on high for 10cycles of 30vortex/30on iceThis step breaks open the yeast by brute force. Why do you think the glass beads are included? Why include cycles on ice?
19) Incubate tubes for 2min on ice.
20) Centrifuge at 95% for 10min.
21) While tubes are spinning- label a clean tube "N" and one clean tube "I"
22) Transfer 900µl of supernatant to correct clean tube.
AVOID ALL DEBRIS at the bottom of the tube as well as 'gunk' layer at the top. It is better to take less than 900ul and avoid 'gunk'. The debris contains membranes, unbroken cells, and proteins. Since the yeast contain cellular RNases (a protein) it is important to separate them from your RNA.
23) Add 800µl isopropanol to each tube. Mix by inverting the tubes for 2 min.
The alcohol reacts with the RNA (which is a salt) and causes it to precipitate out of solution
24) Centrifuge tubes at 95% for 5min to pellet the RNA precipitate.
25) Carefully decant the supernatants by inverting a tube and giving it a decisive flick.
26) Spin the tubes again for 30sec and remove any residual supernatant with a yellow tip and P200 pipet.
27) Resuspend each pellet in 200µl Solution 3. Make sure pellet is resuspended--flick it, vortex it, make sure the pellet is gone.
28) Get Solution 4 from me. Add 40µl to the samples. Mix by vortexing for 10sec.
29) Incubate in your rack at room temp for 5min (return solution 4 to me)
Solution 4 contains a resin that binds DNA but not RNA
30) Spin 90% for 2min.
31) Label a new tube for N and I --include identifying marks since this tube will be stored in the freezer with other samples.
32) Carefully MOVE the SUPERNATANT to the correct clean tube.
Your RNA should always be stored on ice or frozen to slow degradation (why would cold slow degradation?)
Question: What types of RNA have you just isolated? (how many different kinds of RNA are in a cell?) How can RNA analysis help you characterize your 'unknown' strain later in the year? What will it tell you about where your strain is blocked in the mating reaction?
Quantifying RNA
When you perform the dot blot it will be important that each 'dot' contains the same amount of RNA (think about why) Quantification of RNA using spectrophotometry allows you to determine how much RNA you have isolated as well as how free of protein contaminants it is.
(hint: A well-prepared lab group might have 2 members complete this part while the other 2 prepare for the next section)
1) Make sure that spectrophotometer is on.. The UV lamp must be turned on and must have 10min to warm up.
The life of this lamp is GREATLY reduced if you turn it on and off. During this lab just leave it on. When quantifying RNA for your mutants try to coordinate with other groups and leave a note on the spectrophotometer saying when it was turned off. It must have a chance to cool for at least 45min before being turned on again.
2) Get 2 plastic cuvettes from the side bench. Hold cuvettes by the edges--fingerprints on the flat surface will cause inaccurate readings. One cuvette is for each of your samples. A 'blank' cuvette will be left by the spectrophotometer.
3) Place 996µl of sterile distilled water into each cuvette. Add 4µl of your uninduced yeast RNA into one cuvette. Add 4µl of your induced yeast RNA into the other cuvette.
What dilution of RNA did you just perform? A one in __________dilution. This means that you diluted the sample by a factor of ________ (same # as above).
4) Hold a piece of parafilm across the top of the cuvette and mix the contents by inversion.
5) Take cuvettes to spectrophotometer. Set up Spectrophotometer to read absorption at 260nm.
- Press 260 and then the "go to l" key on the key pad
- After numbers in left display stabilize at 260, open lid and place BLANK cuvette (containing water only) in front holder. Flat sides of cuvette must be perpendicular to you.
- Close the lid
- Press the 'Autozero' button (yellow). Wait for right display to stabilize at 0.000
- Open the lid. Remove the blank cuvette and place sample cuvette in its place
- Close the lid. Wait for right display to stabilize--this is the optical density of your RNA sample at 260nm
- Repeat for other RNA sample.
INDUCED sample OD260=__________
UNINDUCED sample OD260=___________
6) Set up Spectrophotometer to read absorption at 280nm. Follow directions as in #5 only press 280 and "go to l" in step A.
INDUCED sample OD280=__________
UNINDUCED sample OD280=___________
7) Calculate the 280/260 ratio. A 'very clean' sample will have a 280/260 ratio of between approximately 0.4 to 0.55
INDUCED 280/260=
UNINDUCED 280/260=
8) Calculate the concentration of RNA in each sample using the following equation.
(Note that the equation only uses the OD260.)
[µg/µl] = [(OD 260) * (Dilution factor)]/24
- bold= conversion coefficient for RNA
- italics= you calculated the dilution factor in #3
INDUCED SAMPLE CONCENTRATION:
UNINDUCED SAMPLE CONCENTRATION:
AFTER you have calculated the sample concentration add 4µl of RNAsin to each sample. RNAsin inhibits RNAses (and keeps your 'RNAs…in…'). This compound should help keep your samples intact.
Isolated RNA (+/- RNAlater from Ambion)
|
MW
|
+later
+Inhib
|
+later
-Inhib
|
-Later
+Inhib
|
-Later
-Inhib
|
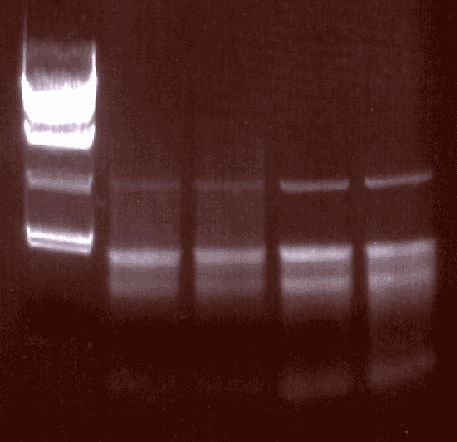
RNA was isloted with and without RNAlater.
Although the yeild was reduced (190 µg v. 240 µg), the quality of the RNA seems to be improved.
RNAsin was also added to compare its utility. No difference was noticed at this time.
Biology Home Page


© Copyright 2001 Department of Biology, Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu