- This web page was produced as an assignment for an undergraduate course at Davidson College -
ICAM-1
also known as CD54
Introduction and Structure
Intercellular Adhesion Molecule - 1 (ICAM-1, CD54) is a transmembrane glycoprotein molecule of the immunoglobulin superfamily. Each molecule is characterized by five distinct immunoglobulin-like domains, a transmembrane domain, and a cytoplasmic tail (Stolpe and Saag, 1996). The entire protein is coded by seven exons and six introns on chromosome 19. Each immunoglobulin domain is coded by a different exon. While the final protein is only 505 amino acids long, the molecule weighs between 80 and 114 kDa depending on the level of glycosylation, which varies among cell types and environments (Newman et al., 1990).
ICAM-1 is a fundamental component in many immune-related processes. ICAM-1 associates with receptors of the integrin family, thereby mediating cell-cell interactions and allowing for signal transduction. ICAM-1 interacts specifically with its receptors to induce a reversible adhesion interaction. Since normal immune function relies on ICAM-1 for processes like T cell activation and leukocyte recruitment, it is understandable that alterations in ICAM-1 structure or expression are associated with immune disorders (Sligh et al., 1993). Therefore it is important to properly understand the various functions and regulatory mechanisms of ICAM-1, the resulting disease-related failures, and the various treatments.
Chime rendering of ICAM-1 (you will need the Chime plug-in to view these images):
N-terminal domain of human intercellular cell adhesion molecule-1 (ICAM-1)
ICAM-1 complexed with human rhinovirus 16
ICAM-1 expression is regulated through four primary pathways: NFkB, JAK/STAT and IFN-g, AP-1 and MAP Kinase, and PKC. Ultimately, ICAM-1 is regulated at the level of transcription by one of these signaling cascades (Roebuck and Finnegan, 1999).
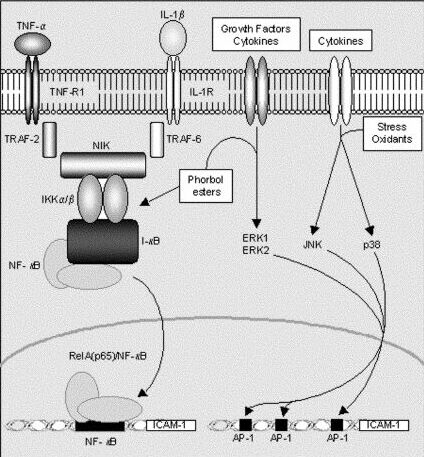
The NFkB pathway is prompted by proinflammatory cytokines such as TNF-a and IL-1b. As shown in figure 1, TNF-a and IL-1b actually activate NIK via different receptors. Regardless, the signal converges with NIK which then degrades IkB. The degraded protein then releases NFkB which translocates to the nucleus and promotes the transcription of ICAM-1. The resultant NFkB response is the most common inducer of ICAM-1 in cells (Shrikant et al., 1994).
Interferon gamma (IFN-g) also has a signaling effect on the transcriptional control of ICAM-1. In the JAK/STAT pathway (Janus kinase/signal transducers and activators of transcription, respectively), JAK elements associate with the cytoplasmic tail of cytokine receptors, cross-phosphorylate, and activate STATs which initiate transcription of genes (Janeway et al., 2001). As figure 2 indicates, STAT homodimers bind an interferon-g response element (IRE) to promote ICAM-1 transcription.
The right side of figure 1 indicates that there are two signaling cascades that can be used to activate the AP-1 promoter of ICAM-1. Unfortunately, the figure is a bit oversimplified, actually both factors are required. Growth factors or cytokines activate ERK1 and ERK2 which then bind to cis elements SRF and Elk-1 which are promoters for the transcription of the transcription factor Fos (Karin, 1996). Meanwhile, other cytokines, oxidants, or stress activate either JNK or p38 signaling cascades which both converge to activate ATF-2 and c-Jun, thereby promoting the transcription of the transcription factor Jun. Fos and Jun then dimerize to make a complete AP-1 binding transcription factor which may associate with one of four upstream locations to promote the transcription of ICAM-1 (Karin et al. 1997).
AP-1 mediated transcription is a more complex pathway than those prompted by TNF-a and IL-1b, but it also affords the cell more control over expression cause by more uncontrollable factors such as stress or oxidants. That is, whereas TNF-a expression is tightly regulated so the response need not be as regulated, less regulated signals such as stress must be better regulated along the signaling cascade (Roebuck and Finnegan, 1999).
Finally, ICAM-1 is indirectly regulated by protein kinase C (PKC). PKC activators such as phorbol ester (figure 1) and phorbol dibutyrate have been shown to increase the expression of ICAM-1. It is assumed that PKC activity is required for general expression of ICAM-1, even via mediators described above such as IL-1b, TNF-a, IFN-g, and other stress factors. The precise mechanism of PKC regulation is not fully understood, but it has been shown that drugs blocking PKC activity inhibit the transcription of ICAM-1 (Rahman et al., 1999).
Figure 1. A brief schematic of ICAM-1 regulatory pathways via NFkB or AP-1 MAPK (Niessen et al., 2002).
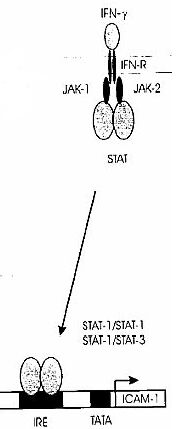
Figure 2. A brief schematic of ICAM-1 regulatory pathway via JAK/STAT and IFN-g (Roebuck and Finnegan, 1999).
The primary receptors for ICAM-1 are integrins which mediate cell-cell interactions and allow for signal transduction. Unlinke most integrin-binding proteins, ICAM-1 does not contain an RGD (Arg-Gly-Asp) motif to promote integrin binding (Stolpe and Saag, 1996). Integrins are characterized by two subunits, a and b, of which there are several a and b families which can combine in different manners to make a wide array of integrins, each with a specific function. Specifically, ICAM-1 is targeted to two integrins of the b2 subunit family: LFA-1 (also aLb2 or CD11a/CD18) and Mac-1 (also aMb2 or CD11b/CD18) (Janeway et al., 2001). The interaction with these two molecules gives ICAM-1 a role in its two most important immune-related functions: T cell function and activation and leukocyte-endothelial cell interaction.
CD8 T cells utilize non-specific interations between ICAM-1 and LFA-1 as the primary step in antigen recognition. The momentary adhesion interaction induced by the ICAM-1 allows the T cell time to align the T cell receptor with the MHC class I:peptide complex. A successful TCR interaction will increase the adhesive force and commence the effector function (Goldstein et al., 2000). Conversely, an unsuccessful TCR interaction will not provide adequate adhesive forces and the T cell will leave the cell and repeat the process elsewhere. Antigen presenting cells (APC) also express ICAM-1 alongside MHC class II. By a mechanism analogous to the CD8-target cell ICAM-1 interaction, APCs also use ICAM-1 to pause CD4 T cells allowing time to prompt activation via interaction of the TCR and the MHC II:peptide complex (Stolpe and Saag, 1996).
The first stage of leukocyte-endothelial interaction is mediated by the weaker non-specific selectin molecules, including P, E, and L selectin. Inflammatory responses will upregulate the expression of ICAM-1 (see above) thereby increasing the adhesive nature of leukocytes and endothelial cells. While the selectins instigate a rolling behavior over the endothelial layer, the ICAM-1 interaction with leukocyte LFA-1 or Mac-1 actually stabilizes the leukocyte for extravasation. The arrested leukocytes then begin diapedesis, the process of crossing the endothelial layer, which is mediated by PECAM (CD31), a protein expressed both on leukocytes and the intercellular junctions of endothelial cells. Though the ICAM-1:integrin interaction is not specific in the same way a TCR is specific for a certain MHC:protein, ICAM-1 is specifically regulated by cytokines and other factors as described above thus controlling the nature of the inflammatory response (Schleimer and Bochner, 1998).
Pathology and Therapy
Mice designed with a knockout of the ICAM-1 gene develop normally but present with high blood neutrophil concentration and decreased capacity for leukocyte activation and migration. These mice therefore also exhibit decreased inflammation reaction and a lesser propensity for septic shock (Sligh et al., 1993). ICAM-1 is also a functional receptor for rhinoviruses (see chime image), which causes some upper respiratory tract infections. Tuncated ICAM-1 has been shown to reduce the viral infection in vitro (Greve et al., 1991).
Defects in ICAM-1 expression have been linked to melanoma, lymphoid malignancies, myeloid malignancies, allergic asthma, atherosclerosis, ischemia, neurological disorders, and organ transplant rejection (Stolpe and Saag, 1996). Of these disorders, there are two approaches to ICAM-1 therapy: antibody-mediated neutralization of ICAM-1 or LFA-1, or pharmacological induction of ICAM-1. One of these opposite treatments are administered depending on the type of disease.
Adhesion blockers are most appropriate for cases where ICAM-1 overexpression is negatively impacting the patient. Injected soluble ICAM-1 (sICAM-1) is not strong enough to outcompete wildtype membrane-bound ICAM-1 (mICAM-1), so sICAM-1 is coupled to an Ig. This dimeric molecule has been shown to block normal LFA-1 to mICAM-1 interactions. Besides blocking the integrin, monoclonal antibodies (mAbs) against ICAM-1 are also used to neutralize the effects of the molecule. This is a commonly used approach used to treat transplant recipients and is being tested as a way to treat allergic asthma and prevent rhinovirus infections (Stolpe and Saag, 1996). Other drugs, such as Zofenopril (Cominacini et al., 2002), have also been shown to inhibit ICAM-1 expression.
Induction of ICAM-1 is promoted in cases where recruitment of cytotoxic T cells is beneficial. Specifically, malignancies both in tissue and in blood that are induced to overexpress ICAM-1 have been shown to grow more slowly than those expressing normal levels of ICAM-1. (Boyer et al., 1995; Okegawa et al, 2002). Though not sufficient to eliminate end-stage cancers, upregulation of ICAM-1 may represent a new treatment option for hematological malignancies in an early phase.
Click the "P" icon next to each periodical reference to view the abstract on PubMed.
![]() Boyer,
MW, PJ Orchard, KB Godem, PM Anderson, RS Mclvor, BR Blazar. (1995).
Dependency on intercellular adhesion molecule recognition and local
interleukin-2 provision in generation of an in vivo CD8(+) T cell immune
response to murine myeloid leukemia. Blood. 85: 2498-506.
Boyer,
MW, PJ Orchard, KB Godem, PM Anderson, RS Mclvor, BR Blazar. (1995).
Dependency on intercellular adhesion molecule recognition and local
interleukin-2 provision in generation of an in vivo CD8(+) T cell immune
response to murine myeloid leukemia. Blood. 85: 2498-506.
![]() Cominacini,
L, A Pasini, U Garbin, S Evangelista, AE Crea, D Tagliacozzi, C Nava, A Davoli,
V LoCascio. (2002).
Zoefenopril inhibits the expression of adhesion molecules on endothelial
cells by reducing reactive oxygen species.
American Journal of Hypertension. 15: 891-5.
Cominacini,
L, A Pasini, U Garbin, S Evangelista, AE Crea, D Tagliacozzi, C Nava, A Davoli,
V LoCascio. (2002).
Zoefenopril inhibits the expression of adhesion molecules on endothelial
cells by reducing reactive oxygen species.
American Journal of Hypertension. 15: 891-5.
![]() Goldstein,
JS, T Chen, E Gubina, RW Pastor, S Kozlowski. (2000). ICAM-1
enhances MHC-peptide activation of CD8(+) T cells without an organized
immunological synapse. European Journal of Immunology. 30:
3266-70.
Goldstein,
JS, T Chen, E Gubina, RW Pastor, S Kozlowski. (2000). ICAM-1
enhances MHC-peptide activation of CD8(+) T cells without an organized
immunological synapse. European Journal of Immunology. 30:
3266-70.
![]() Greve,
JM, CP Forte, CW Marlor, AM Meyer, H Hoover-Litty, D Wunderlich, A
McClelland. (1991). Mechanisms of receptor-mediated rhinovirus
neutralization defined by two soluble forms of ICAM-1. Journal of
Virology. 65: 6015-23.
Greve,
JM, CP Forte, CW Marlor, AM Meyer, H Hoover-Litty, D Wunderlich, A
McClelland. (1991). Mechanisms of receptor-mediated rhinovirus
neutralization defined by two soluble forms of ICAM-1. Journal of
Virology. 65: 6015-23.
Janeway, CA, P Travers, M Walport, MJ Shlomchik. Immunobiology 5. New York: Garland Publishing, 2001.
![]() Karin,
M. (1996). The regulation of AP-1 activity by mitogen-activated
protein kinases. Philosophical Transactions of the Royal Society of
London: Biological Sciences. 351: 127-34.
Karin,
M. (1996). The regulation of AP-1 activity by mitogen-activated
protein kinases. Philosophical Transactions of the Royal Society of
London: Biological Sciences. 351: 127-34.
![]() Karin,
M, Z Liu, E Zandi. (1997). Ap-1 function and regulation. Current
Opinion in Cell Biology. 9: 240-46.
Karin,
M, Z Liu, E Zandi. (1997). Ap-1 function and regulation. Current
Opinion in Cell Biology. 9: 240-46.
![]() Newman,
PJ, MC Berndt, J Gorski, GC White, S Lyman, C Paddock, WA Muller.
(1990). PECAM-1 (CD31) cloning and relation to adhesion molecules of the
immunoglobulin gene superfamily. Science. 247: 1219-222.
Newman,
PJ, MC Berndt, J Gorski, GC White, S Lyman, C Paddock, WA Muller.
(1990). PECAM-1 (CD31) cloning and relation to adhesion molecules of the
immunoglobulin gene superfamily. Science. 247: 1219-222.
![]() Niessen,
HW, PA Krijnen, CA Visser, CJ Meiher, CE Hack. (2002). Intercellular
Adhesion Molecule - 1 in the Heart. Annals of the New York Academy of
Science. 973: 573-85.
Niessen,
HW, PA Krijnen, CA Visser, CJ Meiher, CE Hack. (2002). Intercellular
Adhesion Molecule - 1 in the Heart. Annals of the New York Academy of
Science. 973: 573-85.
![]() Okegawa,
T, Y Li, RC Pong, JT Hsieh. (2002). Cell adhesion proteins as tumor supressors.
Journal of Urology. 167: 1863-43.
Okegawa,
T, Y Li, RC Pong, JT Hsieh. (2002). Cell adhesion proteins as tumor supressors.
Journal of Urology. 167: 1863-43.
![]() Rahman,
A, M Bando, J Kefer, KN Anwar, AB Malik. (1999). Protein kinase - C
activated oxidant generation in endothelial cells signals intercellular adhsion
molecule - 1 gene transcription. Molecular Pharmacology. 55:
575-83.
Rahman,
A, M Bando, J Kefer, KN Anwar, AB Malik. (1999). Protein kinase - C
activated oxidant generation in endothelial cells signals intercellular adhsion
molecule - 1 gene transcription. Molecular Pharmacology. 55:
575-83.
![]() Roebuck, KA and A
Finnegan. (1999). Regulation of intercellular adhesion molecule - 1
(CD54) gene expression. Journal of Leukocyte Biology.
66: 876-88.
Roebuck, KA and A
Finnegan. (1999). Regulation of intercellular adhesion molecule - 1
(CD54) gene expression. Journal of Leukocyte Biology.
66: 876-88.
![]() Schleimer,
RP and BS Bochner. (1998). The role of adhesion molecules in
allergic inflammation and their suitability as targets of antiallergenic
therapy. Clinical Experimental Allergy. 28: 15-23.
Schleimer,
RP and BS Bochner. (1998). The role of adhesion molecules in
allergic inflammation and their suitability as targets of antiallergenic
therapy. Clinical Experimental Allergy. 28: 15-23.
![]() Shrikant, P, IY Chung, ME Ballestas, EN
Benveniste. (1994). Regulation of intercellular adhesion molecule -1
gene expression by tumer necrosis factor - alpha, interleukin - 1 beta,
interferon gamma in astrocytes. Journal of Neuroimmunology.
51: 209-20.
Shrikant, P, IY Chung, ME Ballestas, EN
Benveniste. (1994). Regulation of intercellular adhesion molecule -1
gene expression by tumer necrosis factor - alpha, interleukin - 1 beta,
interferon gamma in astrocytes. Journal of Neuroimmunology.
51: 209-20.
![]() Sligh,
JE, CM Ballantyne, SS Rich, HK Hawkins, CW Smith, A Bradley, AL Beaudet.
(1993). Inflammatory and immune responses are impared in mice deficient in
intercellular adhsion molecule - 1 deficient mice. Proceedings of the
National Academy of Sciences. 90: 8529-33.
Sligh,
JE, CM Ballantyne, SS Rich, HK Hawkins, CW Smith, A Bradley, AL Beaudet.
(1993). Inflammatory and immune responses are impared in mice deficient in
intercellular adhsion molecule - 1 deficient mice. Proceedings of the
National Academy of Sciences. 90: 8529-33.
![]() Stolpe, AV and PT
Saag.
(1996). Intercellular adhesion molecule - 1. Journal of Molecular
Medicine. 74: 13-33.
Stolpe, AV and PT
Saag.
(1996). Intercellular adhesion molecule - 1. Journal of Molecular
Medicine. 74: 13-33.
This
page created by: Gray Lyons ![]()
Return to the Immunology Homepage
Return to the Davidson College Homepage