This web page was produced as an assignment for an undergraduate course at Davidson College
Autoimmune Lymphoproliferative Syndrome
Introduction
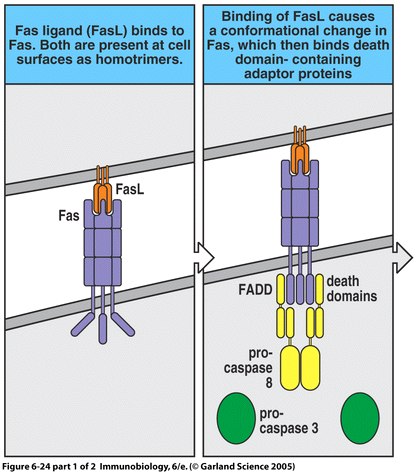
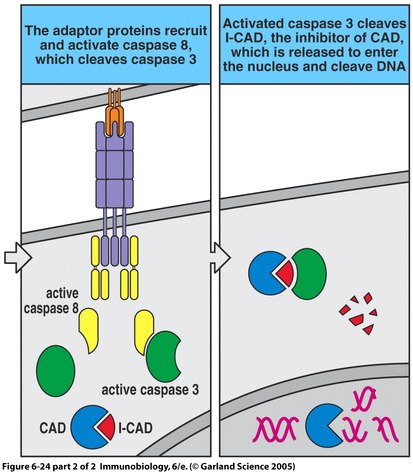
Autoimmune Lymphoproliferative Syndrome (ALPS) is an immune disease resulting from non-functional or dysfunctional apoptosis of lymphocytes (Rao and Straus, 2006), and is characterized by lymphoproliferation, peripheral accumulation of double-negative αβ T cells (DNTs), and disrupted lymphocyte apoptosis (Bidere et al., 2006). This inability to perform proper cell mediated death is caused by several independent pathways that fall into three different classifications. Type I ALPS is subdivided into two categories, Ia and Ib. A patient with type Ia has mutations of the gene encoding Fas, a receptor protein located on the target cell that induces apoptosis. Conversely, type Ib patients have genetic mutations of the Fas ligand on cytotoxic T cells. Type I ALPS is the most prevalent, with Fas and Fas ligand mutations accounting for 75% of reported cases. Type II is much less common and involves the mutation of caspase 8 or caspase 10, which disrupts the caspase cascade that normally results in the cleaving of DNA during apoptosis. Type III ALPS refers to cases in which no mutation can be identified as the cause of apoptosis disruption (Rao and Straus, 2006).
Apoptosis
Apoptosis is described as programmed cell death. In the immune response, apoptosis acts to preserve peripheral T cell homeostasis by regulating responses to foreign antigens; additionally, it maintains tolerance of self-antigens within the body. The elimination of mature, activated T cells after an infection is cleared from the body is important in preventing autoimmune responses (Fisher et al., 1995). ALPS and its disruption of apoptosis allows for accumulation and persistence of activated lymphocytes (Rao and Straus, 2006) that retain the potential for cross-reacting with self-peptides, resulting in an autoimmune response (Fisher et al., 1995).
The majority of ALPS cases stem from the dominant heterozygous mutation of the Fas surface protein (Zhu et al., 2006). Fas is a member of the tumor-necrosis factor (TNF) receptor family (Janeway et al., 2005). It is expressed on activated B and T lymphocytes as well as virally infected cells in the periphery of the body (Rao and Straus, 2006). The binding of Fas to Fas ligand triggers apoptosis in the cell presenting the Fas protein (Janeway et al., 2005); however, the mutation of either of these cell surface proteins disrupts the signaling pathway required for apoptosis (Straus et al., 1999a). Furthermore, for unknown reasons, those patients with mutations of the intracellular portion of Fas (the death domain) are much more likely to present physical symptoms of ALPS (Rao and Straus, 2006).
Figure 1. The apoptosis signaling pathway includes Fas and Fas ligand binding as well as a sequenced caspase cascade.
Effects of ALPS
Individuals affected with ALPS are fully capable of fighting infection of foreign antigens. The problem lies in their ability to effectively scale back the immune response once the antigen is eliminated from the body. Those who are unaffected will kill, through apoptosis, the effector T and B cells once the antigen is disposed of in order to suppress the immune response after it is no longer needed. Those with ALPS are unable to initiate this downgrading of the immune system. The result is a build-up of lymphocytes in the peripheral lymphoid organs (lymph nodes and the spleen) which may reach intolerable levels (Straus et al., 1999b).
Straus et al. (1999a) state that certain features of ALPS have independently been recognized for many years as individual occurrences, but not until recently have they been considered combination to signal the presence of this autoimmune disorder. The first signs of ALPS occur at as early as two months of age of infants (Fisher et al., 1995). The persistence of effector cells in the body after infection, due to non-functional apoptosis, will collect in the peripheral lymphoid organs, leading to lymphadenopathy (enlarged lymph nodes), splenomegaly (enlarged spleen), and eventually cytopenia (reduction in the number of cells circulating in the blood) (Rao and Straus, 2006). Cytopenia is linked to another apparent feature of ALPS development which is the increased prevalence of DNTs in peripheral areas of the body. It is likely these are T cells that once presented either the CD4 or CD8 coreceptor on the cell surface, but have since lost the ability to do so. In unaffected individuals, DNTs comprise less than 1% of all substances in the blood, but can reach up to 40% in those with ALPS (Bidere et al., 2006). The persistence of DNTs in the body may potentially lead to this autoimmune disease because they produce elevated levels of interluekin-10 (IL-10), IL-4, and IL-5, but also decrease IL-2 and IFN-γ levels. This combination of cytokine production and limitation often leads to T-helper 2 cell differentiation, activation of the humoral immune response, and potential autoantibody production. These extra antibodies have the potential to be programmed to recognize red blood cells and platelets, leading to an autoimmune reaction causing cytopenia (Bidere et al., 2006).
ALPS Therapy
There is no specific cure for ALPS. Current treatments involve a variety of approaches, which are tailored to an individual’s particular condition and symptoms. These therapies include the use of corticosteroids such as prednisone or immunosuppressive drugs like cyclosporine (Campagnoli et al., 2006). These drugs suppress the autoimmune response by inhibiting DNA transcription in the cell nucleus. Through this mechanism, they effectively block T-cell proliferation (Janeway et al., 2005). This steroid therapy reduces the number of effector lymphocytes produced by the body; however, it has not been found to consistently decrease the size of the spleen or lymph nodes (Rao and Straus, 2006).
Extreme cases of ALPS resulting in splenomegaly may require surgical treatment to prevent organ rupture as well as cytopenia. A splenectomy is the removal of the spleen which has both positive and negative consequences. This procedure is necessary to protect against splenic rupture and may help in regulating blood cell counts. However, by removing the spleen the patient is losing a major peripheral lymphoid organ involved in the immune response. Furthermore, the patient becomes vulnerable to sepsis following the surgical procedure. It is suggested that a splenectomy only be performed as a last option in controlling splenomegaly and cytopenia (Rao and Straus, 2006).
As previously stated, ALPS often results from the mutation of genes regulating apoptosis, which may lead one to consider gene therapy as a possible form of treatment. Unfortunately, this currently is not a feasible method for preventing ALPS, but should be one that deserves the attention of future researchers and clinicians (Straus et al., 1999b).
References
Bidere, N; Su, HC; Lenardo, MJ (2006) Genetic Disorders of Programmed Cell Death in the Immune System. Annual Review of Immunology. 24, 321-352.
Campagnoli, MF; Garbarini, L; Quarello, P; Garelli, E; Carando, A; Baravalle, V; Doria, A; Biava, A; Chiocchetti, A; Rosolen, A; Dufour, C; Dianzani, U; Ramenghi, U (2006) The Broad Spectrum of Autoimmune Lymphoproliferative Disease: molecular bases, clinical features and long-term follow-up in 31 patients. Haematologica. 91, 106-109.
Fisher, GH; Rosemberg, FJ; Straus, SE; Dale, JK; Middelton, LA; Lin, AY; Strober, W; Lenardo, MJ; Puck, JM (1995) Dominant Interfering Fas Gene Mutation Impairs Apoptosis in a Human Lymphoproliferative Syndrome. Cell. 81, 935-946.
Janeway, CA; Travers, P; Walport, M; Schlomchik, MJ (2005) Immunobiology. Garland Science: New York, NY: 232-233.
Rao, VK and Straus, SE (2006) Causes and Consequences of the Autoimmune Lymphoproliferative Syndrome. Hematology. 11, 15-23.
Straus, SE; Sneller, M; Lenardo, MJ; Puck, JM; Strober, W (1999a) An Inherited Disorder of Lymphoproliferative Apoptosis: The Autoimmune Lymphoproliferative Syndrome. Annals of Internal Medicine. 130, 591-601.
Straus, SE; Sneller, M; Dale, JK; Puck, JM; Lenardo, MJ (1999b) Autoimmune Lymphoproliferative Syndrome. <http://www.niaid.nih.gov/Publications/alps/pdf/alps.pdf>. Accessed 2006 Apr 24.
Zhu, S; Hsu, AP; Vacek, MM; Zheng, L; Schaffer, AA; Dale, JK; Davis, J; Fischer, RE; Straus, SE; Boruchov, D; Saulsbury, FT; Lenardo, MJ; Puck, JM (2006) Genetic Alterations in Caspase-10 May be Causative or Protective in Autoimmune Lymphoproliferative Syndrome. Human Genetics. 119, 284-294.