 |
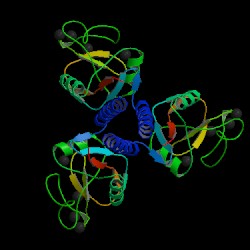 |
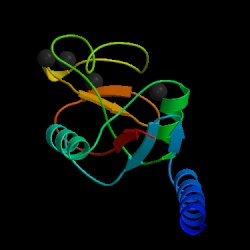
Fig. 1a MBL subunit |
Fig. 2 MBL trimer Source: Protein Data Bank, 2006 |
Fig. 1b JMOL image of MBL subunit
Source: Protein Data Bank, 2006
Mannose-binding lectin (MBL), also called mannose binding protein (MBP), is a calcium-dependent serum protein that plays a role in the innate immune response by binding to carbohydrates on the surface of a wide range of pathogens (viruses, bacteria, fungi, protozoa ) where it can activate the complement system or act directly as an opsonin (Anders, 2001). Mannose-binding lectin is a member of the collectin family of proteins, which are made in the liver and can opsonize bacteria by tagging the surface of a pathogen to facilitate recognition and ingestion by phagocytes. Collectins get their name because they have a collagen-like region and a lectin region. Lectins are proteins that bind sugar molecules, usually on the surface of bacteria. The collagen domain interacts with the effector parts of the innate immune system. The MBL2 gene on human chromosome 10 produces MBL, an oligomer of 248-amino acid protein subunits composed of three identical polypeptide chains comprising a cistine rich region, a collagen-like region, a neck, and a carbohydrate recognition domain (Matsushita, et al, 2000). Three MBL polypeptide chains assemble into a biologically active trimer found in vivo.
|
|
Fig. 1a MBL subunit |
Fig. 2 MBL trimer Source: Protein Data Bank, 2006 |
Fig. 1b JMOL image of MBL subunit
Source: Protein Data Bank, 2006
The complement system is a set of plasma proteins that work together to attack extracelluar pathogens. While the most important role of the complement system is to opsonize pathogens, it also recruits inflammatory cells and kills pathogens directly through membrane attack complexes. The complement system can be activated through three pathways: the classical pathway, the alternative pathway, and the mannose-binding (MB) lectin pathway (see Fig 4 below).
One of the biggest obstacles to making sense of the complement system is understanding the nomenclature of the proteins involved. All components of the classical complement pathway and membrane attack complex are designated by the letter C followed by a number. What is confusing is that the components were numbered in order of their discovery rather than the sequence of reactions. The reaction sequence is C1, C4, C3, C5, C6, C7, C8, and C9. The product of each cleavage reaction are designated by added lower case letters, the larger fragment is called ‘b’ (a helpful mnemonic is ‘b’ stands for big) and a smaller fragment called ‘a.’ The three complement pathways follow a different sequence of “early” cleavage reactions that all lead to the assembly of a protease named C3 convertase. C3 is a protein involved in complement which is cleaved by the C3 convertase into two fragments, C3a and C3b. C3a is a mediator of inflammation while C3b covalently binds to the pathogen surface, coating it and acting as an opsonin. The major proteins involved in complement can be summarized according to their function: (Janeway et. al., 2005)
1. Opsonisation: C3b and, to a lesser degree, C4b molecules are opsonins. They coat foreign organisms, greatly enhancing their phagocytosis because phagocytes have receptors that recognize complement proteins bound to pathogen.
2. Inflammation: The C5a and, less potently, the C4a and C3a fragments are important inflammatory activators inducing vascular permeability, recruitment and activation of phagocytes.
3. Lysis: C5b binds and recruits C6 and C7 to the target surface. C7 and subsequently C8 change conformation to expose hydrophobic domains which insert in the lipid bilayer. The C5b678 complex catalyses the polymerisation of the final component C9 which forms a transmembrane pore of ~ 10nm diameter causing lysis of the cell. This macromolecular assembly is known as theMembrane Attack Complex (MAC).
4. Immune complex clearance: Complement has a very important role in solubilising and causing removal from the circulation of immune complexes. It does this by the binding of C4b and C3b, covalently bound to the immune complex, to CR1 complement receptors on red blood cells which transport the complexes to the liver and spleen where they give the complexes up to phagocytes for destruction.
Complement proteins start to be made in the first trimester of fetal development, and are an important part of innate immunity (Sompayrac, 1999). Although the innate immune system is not as specific as adaptive immunity, it can distinguish self from nonself via receptors that recognize patterns of repeating structural motifs on the surface of microorganisms. Pathogens have surface sugars, like mannose, which are absent from mammalian cell surfaces. MBL is an example of a pattern-recognition receptor present as a free protein in blood plasma. MBL binds to bacterial surfaces that display a particular spatial arrangement of carbohydrate sugar residues, mannose or fructose (Janeway et al., 2005).
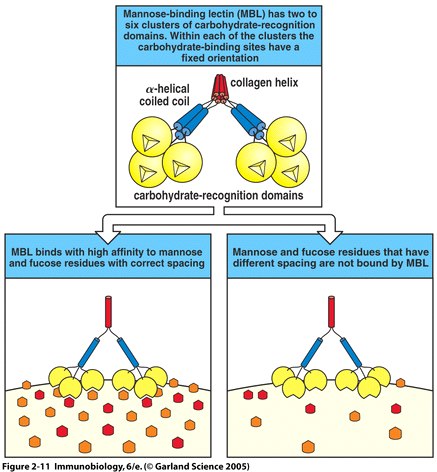
Fig. 3 MBL recognizes bacterial surfaces by their particular spacing of carbohydrate residues (Fig. 2.11 from Janeway et al. , 2005) .
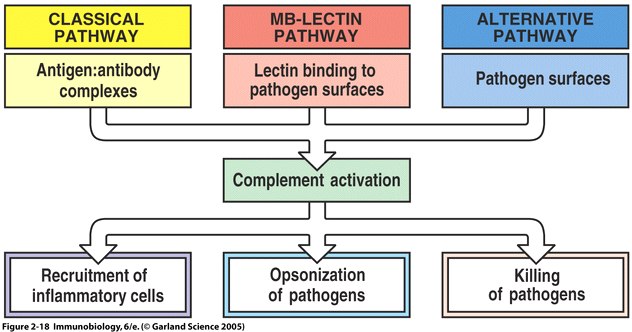
Fig 4 The main fuctions of complement are recruitment of inflammatory cells, opsonization of pathogens, and killing of pathogens
Once MBL recognizes a pathogen, its lectin domain will bind to mannose, or other carbohyrate sugar residues on the pathogen surface, and activate complement via the MB-lectin pathway ( Fig. 2.18 from Janeway et al. , 2005 ).
The complement system can be activated through three pathways: the classical pathway, the alternative pathway, and the mannose-binding (MB) lectin pathway (see Fig 4 above). The most-recently discovered mannose-binding lectin pathway activates complement through the mannose-binding lectin protein. MBL binds to carbohydrates found on the surface of many pathogens. For example, MBL has been show to bind to yeasts such as Candida albicans , to viruses such as HIV and influenza A, to many bacteria including Salmonella and Streptococci , and to parasites like Leishmania (Sompayrac, 1999).
In order to active the complement system, MBL in the blood complexes with (binds to) another protein, a serine protease called MASPs (MBL-associated serine proteases). When MBL binds to its target (for example, mannose on the surface of a bacterium), the MASP protein functions like a convertase to clip C3 into C3a and C3b. C3 is abundant in the blood, so this happens very efficiently. The C3b fragments can then bind to the surface of the bacterium. C3b can cause the complement cascade can proceed in combining with other complement proteins to make a membrane attack complex, which causes lysis of pathogens and cells. C3b can also bind to complement receptors on phagocytes causing opsonization of pathogens (Somapayrac, 1999).
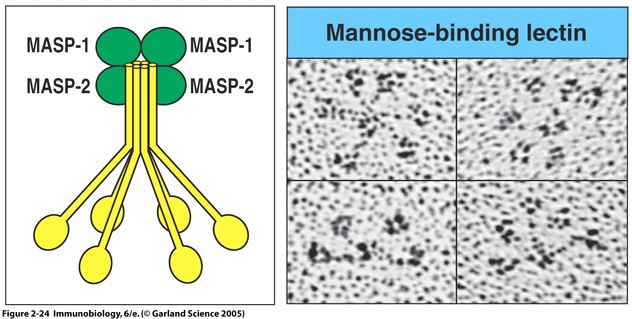
Fig. 5 MBL forms a complex with serine proteases .
Right panel: MBL forms clusters of two to six carbohydrate-binding heads around a central collagen-like stalk and complexes with MBL-associated serine proteases 1 (MASP-1) and 2 (MAPS-2). On binding of MBL to bacterial surfaces, these serine proteases become activated and can then activate the complement system by cleaving and activating C4 and C2. Left Panel: Electron microscope photograph of a MBL cluster. The structure has been described as looking like a bunch of tulips. The central collagen-like stalk is the flower stem and the MBL head is the flower. ( Fig. 2.24 from Janeway et al. , 2005).
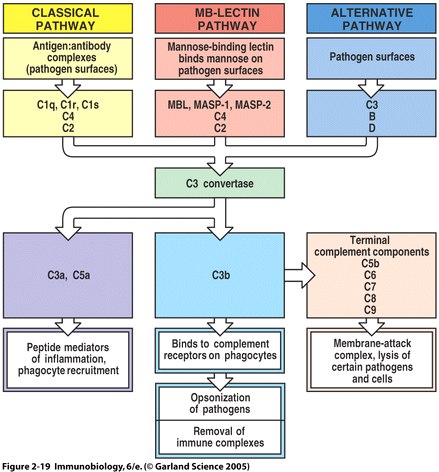
Fig. 6 Overview of the main components and effector actions of complement
Note that the MBL pathway involves the MBL protein, MASP-1, MASP-2, C4, and C2. MASP acts as a C3 convertase, creating a C3b fragment from C3. C3b attaches to the pathogen surface and binds to receptors on phagocytes leading to opsonization. C3b can also combine with other proteins on the pathogen surface and form a membrane attack complex ( Fig. 2.19 from Janeway et al. , 2005 ).
The MBL2 gene encodes mannose-binding lectin (MBL) that is secreted by the liver into the bloodstream (Ohlenschlaeger et al., 2004). Although serum levels of MBL are normally rather low (1500 micrograms/litre), MBL has a crucial role in innate immunity (Turner, 1998). The frequency of this deficiency due to mutations of the MBL2 gene in the general population has been estimated to be between 5 and 10% ( Turner, 1991 ). MBL deficiency arising from mutations and promoter polymorphisms in the MBL2 has been associated with increased risk, severity, and frequency of infections and autoimmunity (Flemming, et. al., 2004).
Although most individuals with MBL deficiency are healthy, they have an increased susceptibility to certain illnesses. The deficiency has been reported to be particularly common in infants with recurrent respiratory tract infection, otitis media, and chronic diarrhea. The low levels of MBL in young children with recurrent infections suggest that the MB-lectin pathway is important during the interval between the loss of passively acquired maternal antibody and the acquisition of a mature immunologic repertoire (Summerfield et al., 1997).
MBL deficiency is also associated with non-infectious diseases in adults including systemic lupus erythematosus, rheumatoid arthritis, cystic fibrosis and common variable immunodeficiency. MBL deficiency increases susceptibility to infectious disease. One study that found a significantly higher number of HIV-infected homosexual males were homozygous for variant MBL2 alleles than were high-risk homosexual controls or healthy controls. Although no significant association was found in progression from infection to clinical AIDS, there was a significantly shorter mean survival time after AIDS diagnosis in men carrying variant MBL2 alleles and those with low serum MBL. The increased risks may be associated with increased susceptibility to coinfections (Garred et.al, 1997). MBL-deficiency also poses a problem for other immuno-compromised individuals, such as cancer patients undergoing chemotherapy. In these patients, there was a strong correlation between MBL-deficiency and the occurrence of clinically significant infections. In addition, studies of cystic fibrosis patients found that MBL-deficient patients have a life expectancy 8-years shorter than MBL sufficient individuals, due to increased bacterial colonization of the lung (Trevisiol, 2005).
If MBL is involved in clearing away bacteria and other pathogens, why is MBL deficiency associated with autoimmune diseases such as lupus and rheumatoid arthritis? The immune system has redundant pathways that all have the same function so that if one is not working well, the immune system can still operate. For example, the classical, MB-lectin, and alternative pathway all make a C3 convertase which leads to oposinzation of pathogens, recruitment of inflammatory cells, and killing of pathogens (see Fig. 4). If the MB-lectin pathway is not working well due to MBL deficiency, the immune system compensates by increasing activation via the other pathways. Most notable is the increase in antibody concentration, used in the classical pathway. Studies have shown an increase in serum levels of IgM antibody concentrations in MBL deficient patients with rheumatoid arthritis. The more antibody, the higher the chance that some antibody will be self-reactive, which is what happens in arthritis( Jacobsen, 2001).
It is also interesting to note the effect of MBL gene polymorphisms on the susceptibility to infection after liver transplantation because all MBL is hepatically produced. Infection is the primary cause of death after liver transplantation. As transplant patients require immune suppressive drugs in order to insure graft survival, they rely to a great extent on their innate immunity to counteract infections. Studies show the presence of MBL variant alleles in the MBL gene of the donor liver, but not in the recipient, is associated with a strongly increased incidence of clinically significant infections following transplantation in a gene-dose-dependent way. This confirms that serum MBL is produced by the liver and is under strong genetic control. The ability to identify a group of patients severely prone to infection post transplantation is of significant clinical value. In an era of donor supply, donor selection based upon MBL genotype is inconceivable. Patients receiving an MBL-variant liver could benefit from MBL replacement therapy similar to that presently being studied in phase I/II and III clinical studies (Bouwman, 2003).
MBL therapy is an area of current research. Therapy for MBL deficient patients involves giving an intravenous infusion of purified mannose-binding lectin obtained from human donor plasma in an attempt to prevent or ameliorate infections. MBL therapy could be used in three clinical scenarios. Firstly, where MBL deficiency leads to increased susceptibility to disease, MBL replacement could be used to increase resistance to that disease. Secondly, in an acute infection MBL therapy might, by enhancing the immune response, speed the resolution of disease in MBL-deficient patients. However, by analogy with complement deficiencies, a potential hazard of MBL replacement in infection is that it might actually do more harm than good by increasing complement-mediated host damage. Uncontrolled activation of the complement system through the MBL pathway may cause inflammation resulting in tissue damage. Thirdly, MBL therapy could be used to alter the natural history of chronic diseases (Summerfiled, 2003).
The usefulness of replacement MBL therapy has shown promise in enhancing recovery to bacterial disease. It will take many years and larger numbers of patients to detect significant changes in the natural history of MBL deficiency associated chronic diseases like rheumatoid arthritis. One study followed a cystic fibrosis patient given MBL pooled from human plasma as an intravenous infusion twice a week for a period of 3 months. The patients's clinical condition was stabilized during the treatment period, but was not improved(Garred, et. al, 2002). Future study of MBL therapy should provide more definitive answers to the effectiveness of short-term and long-term therapy.
No interactions between MBL and drugs were listed in OMIM.
Anders, Koch et.al. (2001) Acute Respiratory Tract Infections and Mannose-Binding Lectin Insufficiency During Early Childhood. JAMA. 285:1316-1321.
Bouwman, Lee et. al. (2003) Mannose Binding Lectin Gene Poymorphisms Confer a Major Risk for Severe Infections After Liver Transplantation. Gastroenterology. 129 (2): 408-414.
Garred, et. al. (2002) MBL therapy in an MBL-deficient patient with severe cystic fibrosis lung disease. Pediatric Pumonology. 33 (3):2001-2007.
Flemming et.al. (2004) Disease-associated Mutations in Human Mannose-binding Lectin Compromise Oligomerization and Activity of the Final Protein. Journal of Biological Chemistry. 279: 21302-21311.
Jacobsen, S et. al. (2001)The Influence of mannose binding lectin polymorphisms on disease outcome in early polyarthritis. Journal of Rheumatology. 28(5): 935-42.
Janeway, et. al. Immunobiology. Garland Science: New York, NY. 2005. 50-61.
Matsushita, Misao et al. (2000) Proteolytic Activities of Two Types of Mannose-Binding Lectin-Associated Serine Protease. The Journal of Immunology.165: 2637-2642.
Ohlenschlaeger, T. et. al. (2004) Mannose-binding lectin variant alleles and the risk of arterial thrombosis in systemic lupus erythematosus. New Eng. J. Med. 351: 260-267.
OMIM: http://www.ncbi.nlm.nih.gov/omim/ . Accessed on March 10, 2006.
Protein Data Bank <http://www.pdb.org> Accessed on March 13, 2006.
Sompayrac, Lauren. How the Immune System Works . Blackwell Science: Malden, MA. 1999. 17-19.
Suankratay, M. et. al. (1999) Mechanism of complement-dependent haemolysis via the lectin pathway: role of the complement regulatory proteins. Clinical and Experimental Immunology. 117: 442.M
Summerfiled, JA., Clinical potential of mannose-binding lectin-replacement therapy. Biochemisty Society Transactions. Aug. 2003: 770-3.
Summerfield, et.al. (1997) Association of mutations in mannose binding protein gene with childhood infection in consecutive hospital series. Brittish Medical Journal 314: 1229-1231.
Trevisiol, C. (2005) MBL2 polymorphisms screening in a regional Italian CF Center. Journal of Cyctic Fibrosis. 4(3): 189-91.
Turner, M. W. (1991) Deficiency of mannan binding protein--a new complement deficiency syndrome. Clin. Exp. Immunology. 86: 53-56.
Turner, M.W . (1998) Mannose-binding lectin (MBL) in health and disease. Immunobiology. 199 (2): 327-39.
Return to my Immunology Homepage
Please send questions, comments, suggestions to camohr@davidson.edu
Copyright 2006 Carolyn Mohr and Davidson College