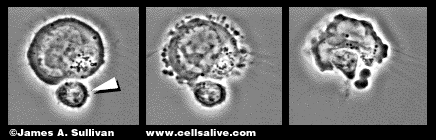
Permission to include this image of a cytotoxic T lymphocyte granted
by
James Sullivan. Please visit his web page at www.cellsalive.com.
Please
click on the image to see a movie of a cytotoxic T cell killing
a target cell..
PERFORIN
Perforin is a 60-kDa cytotoxic protein (Spaner et al. 1999), which is stored in lytic granules on the surface of cytotoxic T cells. When a cytotoxic T-cell receptor recognizes antigen on the surface of a target (i.e infected) cell, perforin as well as other cytotoxic effector proteins, are released by local exocytosis and induce the target cell to undergo apoptosis (Spaner et al. 1999; Alberts et al. 1994).
APOPTOSIS
Cells can die in either of two ways.
First, they can die due to physical or chemical injury or to membrane damage,
which leads to necrosis or cell disintegration. The dead tissue is then
taken up and degraded by phagocytic cells. Second, cells may undergo programmed
cell death, also referred to as apoptosis, in which the cellular DNA is
broken down into 200 base pair fragments and the cell is destroyed from
within (Janeway et al., 1999). Neighboring phagocytic cells ingest the
cell rapidly and prevent the release of cytosolic contents into the extracellular
space. Thus, no inflammatory response is elicited and the DNA of the engulfed
cell can be reused. For this reason, apoptosis is often referred to as
the quiet death (Alberts et al. 1994).
Cytotoxic T cells kill infected cells
by inducing them to undergo apoptosis. The two main pathways utilized by
cytotoxic T cells are the perforin/granzyme pathway and the Fas/Fas-L pathway
(Figures a and b). There are, however, slower pathways such as those mediated
by lymphotoxin and tumor necrosis factor (TNF), which are also involved
in cytotoxic T-cell induced apoptosis (Spielman et al., 1998).
Permission to include this image of a cytotoxic T lymphocyte granted
by
James Sullivan. Please visit his web page at www.cellsalive.com.
Please
click on the image to see a movie of a cytotoxic T cell killing
a target cell..
THE PERFORIN/GRANZYME PATHWAY
When a cytotoxic T cell recognizes
antigen on the surface of a target cell, it releases the contents of its
lytic granules through a calcium-dependent process. These granules contain
two major classes of cytotoxic effector proteins- perforin and proteases
known as granzymes (Janeway et al., 1999). Perforin is released through
exocytosis at the point of contact (Alberts et al., 1994) and polymerizes
within the membrane of the target cell (Spaner et al., 1999), producing
a cylindrical structure in the lipid bilayer that is lipophilic on the
outside and hydrophilic down the length of its hollow center. Water and
salts are then able to enter the cell through these pores, destroying the
integrity of the target cell membrane (Janeway et al., 1999).
In addition to water and salts, granzymes (specifically
granzyme A and B), which have also been released from the lytic granules,
can now enter the target cell. In vitro experiments indicate that the perforin-dependent
entry of granzymes precedes the DNA fragmentation and breakdown of the
nuclear envelope associated with apoptosis.(Blink et al., 1999). More specifically,
granzyme A and B are introduced into the target cell and activate the caspase
family of proteases (Hashimoto et al., 2000). The caspase cascade eventually
leads to the activation of caspase-activatable DNase (CAD) which can then
enter the nucleus of the target cell and cleave the DNA into 200 base pair
fragments. Prevention of granzyme translocation through treatment of target
cells with caspase inhibitors, blocks these apoptotic events (Blink et
al., 1999).
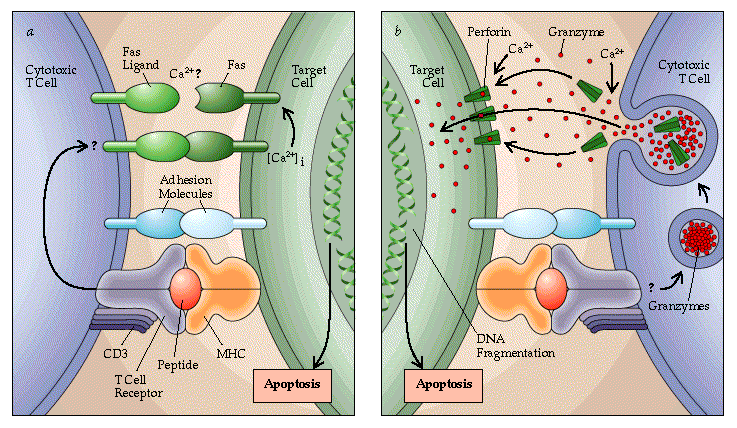
Received permission to use this figure from Scientific
American. Please visit there website at www.sciam.com
(a) apoptosis through nonsecretory Fas-Fas ligand interaction
(b) apoptosis through secretory mechanisms
Gene Knock-Out
Mice that are perforin/Fas-L double-deficient
suffer from severe autoimmune disease, indicating an interesting role for
these two cytotoxic pathways in the regulation of cell-mediated tissue
destruction. It appears that each pathway alone is capable of effectively
regulating this type of tissue destruction, since neither perforin nor
Fas-L single-deficiency produced a similar syndrome. The maintenance of
homeostasis in the immune system is a novel function for perforin and one
which seems to occur only in the absence of Fas-L. Spielman et al. hypothesize
that cytotoxic T cells control their own down-regulation through a negative-feedback
loop, in which they lyse the APCs containing the original antigenic stimulus.
Continued survival of the APCs in the presence of activated T cells may
result in constant restimulation and proliferation, eventually leading
to severe tissue destruction by mechanisms independent of the perforin-
and Fas-L-pathways. Thus, the negative-feedback loop is eliminated in perforin/Fas-L
double-deficient mice and autoimmunity ensues (Spielman et al., 1998).
These findings are supported by other
recent data concerning the induction of vascular leak syndrome (VLS). Cancer
patients treated with high doses of IL-2 suffer significant damage to their
endothelial cells which eventually leads to toxicity characterized by dypsnea,
ascites, weight gain and pulmonary edema. This toxicity is caused by VLS
which involves increased capillary leak. Rafi et al. found that IL-2 upregulates
the activity of perforin and Fas-L. In perforin knockout mice, there was
no significant damage to endothelial cells and VLS was notably reduced.
Fas-L does not appear to play a significant role in tissue damage within
the lungs as Fas-L deficient mice exhibited no decrease in VLS in this
area. However, perforin knockout mice exhibited a marked decrease in VLS
in the lungs. Thus, though IL-2 has proven effective in the treatment of
certain types of cancer, there must also be therapeutic intervention during
treatment to prevent endothelial cell damage (Rafi et al., 1998).
Mice who have had the perforin gene
knocked-out are severely impaired in their ability to mount effective cytotoxic
T cell responses to many, but not all, viruses. Alternatively, mice that
are defective in the gene for granzyme B suffer a less profound deficit.
This is most likely due to the fact that there are several genes encoding
for
ganzymes (Janeway et al., 1999).
The action of cytotoxic T cells mediated
by perforin- or Fas ligand dependent mechanisms are essential for donor
T cells to prevent allogeneic bone marrow rejection. A recent study found
that the ability to prevent graft rejection in mice was severely impaired
by the absence of perforin and was completely eliminated in double mutants
that had perforin-deficient and Fas-ligand-defective CD8 cells (Clarke,
1998).
Other Recent Studies on Perforin
A recent study demonstrated the effect
of perforin-mediated cytotoxic T cell function in neurological disease.
Mice that were deficient for perforin were injected with Theiler's Murine
Encephalomyelitis Virus (TMEW) (model for Multiple Sclerosis). These mice
showed viral persistence in the central nervous system, demyelination in
the white matter of the spinal cord, and chronic brain pathology. However,
these mice revealed only minimal neurological defects as a result of demyelination.
Mice with functional CD8+ T-cells and perforin molecules suffered comparable
demyelination, but had severe clinical disease. Thus, this study indicates
that perforin release by CD8+ T-cells may contribute to the induction of
neurological disease following demyelination (Murray et al., 1998).
Perforin has also been implicated
in the pathogenesis of acute lung injury. Quantitative polymerase chain
reaction (PCR) analysis revealed that the mRNAs for perforin were highly
upregulated in the acute phase of acute respiratory distress syndrome (ARDS)
following sepsis. This finding combined with other results suggest that
the dual apoptosis pathway (perforin/granzyme) (Fas-ligand/Fas) is a likely
contributor to the accumulation and activation of inflammatory cells in
the lungs (Hashimoto et al., 2000).
It appears that perforin/Fas-ligand
double deficiency may also contribute to the expansion of macrophages,
pancreatitis and, in females, infertility and hysterosalpingitis. Double
deficient CD4 or CD8 cytotoxic T-cells are unable to lyse cognate-activated
macrophages. Thus, they cannot mediate negative feedback regulation by
lysis of antigen presenting cells and T cells continue to be activated.
These findings provide insight into a possible homeostatic role for perforin
in the immune system (Spielman, 1998).
WORKS CITED
Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K., Watson, J.
Molecular Biology of the Cell, 3rd ed. New York:
Garland Publishing, Inc., 1994.
Blink, E., Trapani, J., & Jans, D. 1999. Perforin-dependent nuclear
targeting of granzymes: A central role in the nuclear events
of granule-exocytosis-mediated apoptosis? Immunological
Cell Biology 77(3): 206-215.
Clarke, C. 1999. Absence of Perforin Affects Ability to Prevent Rejection. Blood Weekly: 1.
Hashimoto, S., Kobayashi, A., Kooguchi, K., Kitamura, Y., Onodera, H.,
Nakajima, H. 2000. Upregulation of two death
pathways of Perforin/Granzyme and FasL/Fas in septic
acute respiratory distress syndrome. American Journal of
Respiratory Critical Care Medicine 161(1):
237-243.
Janeway, Charles A. Jr., Paul Travers, Mark Walport, and J. Donald Capra.
ImmunoBiology: The immune system in health
and disease, 4th ed. London: Elsevier Science, 1999.
Murray, P., McGavern, D., Lin, X., Njenga, M., Leibowitz, J., Pease,
L., Rodriguez, M. 1998. Perforin-Dependent
Neurologic Injury In A Viral Model of Multiple Sclerosis.
Journal
of Neuroscience 18: 7306-7314.
Rafi, A., Zeytun, A., Bradley, M., Sponenberg, D., Grayson, R., Nagarkatti,
M., & Nagarkatti, P. 1998. Evidence for the
Involvement of Fas Ligand and Perforin in the Induction
of Vascular Leak Syndrome. The Journal of Immunology
161(6):3077-86.
Spaner, D., Raju, K., Rabinovich, B., & Miller, R. 1999. A Role
for Perforin in Activation-Induced T Cell Death in Vivo:
Increased Expansion of Allogeneic Perforin-Deficient
T Cells in SCID mice. The Journal of Immunology 162(2):1192-9.
Spielman, J., Lee, R., & Podack, E. 1998. Perforin/Fas-Ligand Double
Deficiency is Associated with Macrophage Expansion
and Severe Pancreatitis. The Journal of Immunology
161: 7063-7070.
Link to the Immunology Home Page
Link to Davidson College Home Page
© Copyright 2000. Davidson College Biology Department, Davidson College, Davidson, NC, 28036