This molecular analysis of the protein myosin was done using the MacDNAsis program. Below are a series of figures and associated text that analize the DNA sequence that encodes myosin and tries to predict its hydrophobic portions, the sites that would be ideal for the binding of an antibody, and its secondary structure. Also, This program takes the five similar amino acid sequences from the yeast, c.elegans, homo sapiens, chicken and mouse and creates a phylogenetic tree showing the degree of amino acid conservation between these five diverse organisms.
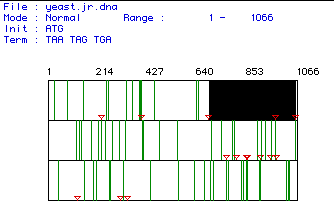
Figure 1. Yeast DNA Analysis
This figure shows a search for the open reading frame (ORF) of the gene encoding the yeast myosin protein. Each of the three stacks represents a separate search for the open reading frame. The green lines represent stop codons, while the red triangles are start codons. The black rectangle, found between nucleotides 640 and 1065, represents the largest ORF and should encode the amino acids responsible for translating the myosin, but this ORF seems too small. The molecular weight of the translated protein was determined to be 1471 Daltons, an amino acid sequence too large to be associated with an ORF of 425 nucteotides. With a molecular weight of 1471 Daltons, we would expect the ORF to be around 4323 bp, some ten times more than the ORF shown above. Therefore, the nucleotide sequence above is probably not cDNA, but instead the complete gene, including exons as well as introns. To see the complete sequence of the gene encoding myosin in yeast, click here: Yeast (S.cereviciae) gene
Figure 2. Kyte and Doolittle hydrophobicity plot of Yeast Myosin
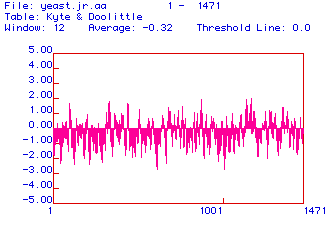
This figure is Kyte and Doolittle hydrophobicity plot of the yeast myosin protein with a window of 12 amino acids. This plot tries to predict whether myosin is an integral transmembrane protein. Amino acid residues 1-1471 are represented on the X-axis, while the hydrophobicity index is represented on the Y-axis.The threshold line is at 0.0: any part of the bar graph above this threshold represents a hyrdophobic portion of the protein, while any part of the bar graph below the threshold line represents a hydropilic portion of the protein. The hydrophilic portions are composed of polar, electrically charged amino acids, while the hydrophobic portions are composed of non polar amino acids. To predict if myosin is a transmembrane protein, we need to investigate the hydrophobic portions of the protein. The critical value on the hydrophobicity index is around 1.8. Any bar that reaches or goes above this value suggests a transmembrane segment. This figure does not show many potential transmembrane segments. Segments around amino acids 900 and 1200 might reach 1.8, but seem to fail to climb above 2.0, so there is no strong evidence suggesting that yeast myosin is a transmembrane protein.
Figure 3. Hopp and Woods Hydropathy Plot
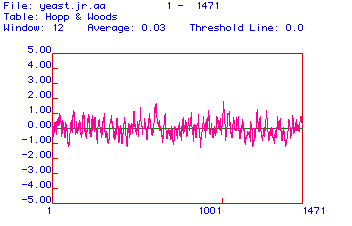
This figure is a Hopp and Woods hydropathy plot of the yeast myosin protein with a window of 12 amino acids.The Hopp and Woods plots try to predict the hydrophilic portions; therefore, the parts that stand the highest are the more hydrophilic segments, and are more likely antigenic. In other words, the highest peaks represent amino acids that correspond to the parts of the protein that are not burried within the folds of the protein or membranes, and are therefore likely spots for an epitope binding site for an antibody. By looking at this figure, it appears that amino acids around 600 and 1000 would be likely spots for mAB recognition and binding. Therefore, these sections should be used to generate a peptide if you need to make a mAB against the peptide for the purpose of probing or isolating the myosin protien. The mAB could more easily recognize a linear epitope from the segments around amino acids 600 and 1000 while the protein is in its linear conformation.
Figure 4. Predicted Secondary Structure of the Yeast Myosin

This figure shows the predicted secondary structure of yeast myosin. These predictions are based on similar amino acid sequences and their effect on a known protein's secondary structure, not on real data. These predictions show predicted helical structures (H), pleated sheets (S), turns (T) and coils (C). We see many long helical structures (between amino acids 29-40, 196-220 and 327-350 for instance), many pleated sheets (between amino acids 1-11, 96-110and 118-130 for instance), three turns and a few coils. This predicted secondary structure must be looked at in comparison to the actual image produced by data as seen in the RasMol image of yeast myosin. The observed secondary structure does show many helical structures on both globular structures, as well as the elongated protein strand connecting the two globular structures. Here, the predicted secondary structure seems to agree with the actual secondary structure. However, when looking at the RasMol image, there is only one pleated sheet in one of the globular structures. The computer predictions showed at least three pleated sheets, so in this instance, the predicted secondary structure differed from the actual. The computer prediction olny predicted one segment of coils, but this segment was small. The RasMol image shows no coils. The take home message from this figure is as follows: Although computer predictions can be a valuable tool in obtaining an image of a protein's secondary structure, these predictions should not be the only images consulted. As we see in the above figure and the RaMol image of myosin, the actual structure can differ significantly from the predicted. One should be cautious in interpreting computer generated images not based on actual data.
Figure 5. Phylogenetic Tree
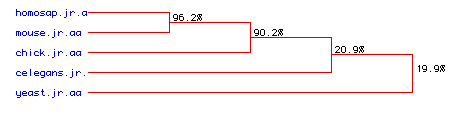
This figure shows a phylogenetic tree that investigates the degree of amino acid conservation over time between the proteins of five diverse organisms. Similar amino acid sequences from homo sapiens, mouse, chicken, c.elegans and yeast were compared, and the % similarities are shown at the right. The amino acid sequences of homo sapiens and the mouse are 96.2% similar, suggesting a high degree of amino acid conservation between these two organisms' myosin proteins. Homo sapiens and mouse also share a high degree of similarity with chicken amino acids encoding myosin, 90.2%, suggesting that all three of these organisms share a high degree of amino acid conservation. C. elegans and yeast show a low degree of similarity to eachother and to the rest of the organisms. These results make sense, for it is expected that homo sapiens , mouse and chicken would have a high degree of amino acid conservation, for these three organisms are more closely related to eachother than the c. elegans and yeast. If the molecular clock ticks, it would be expected that more closely related organisms would show less amino acid changes or substitutions. Therefore, this phylogenetic tree is consistent with what we expected. To see an example of amino acid conservation and substitution, see Figure 6.
To see the amino acid sequences of the five organisms' myosin proteins, click on one of the following:
Figure 6. Amino acid alignments

This figure shows the amino acid alignments of the five organisms of comparison. The black boxes indicate identical amino acid residues, while the white boxes indicate a substitution of amino acids. Dashes indicate shifts in amino acid sequences for the purposes of alignment. The top two panels (amino acids 251-350) show a relatively high degree of amino acid similarity between all five organisms, but especially between the chicken, homo sapiens and mouse. The bottom panel (amino acids 351-400) shows many differences in amino acid residues, especially for the yeast and c. elegans. These amino acid alignments, from amino acids 251 to 400, represent a sample of the alignments used to generate the phylogenetic tree in Figure 5.
To see the amino acid sequences of the five organisms' myosin proteins, click on the following: