This page was produced as an assignment for an undergraduate course at Davidson College.
How to Use NCBI Blast
The National Center for Biotechnology Information (NCBI) maintains a molecular biology public database and develops software tools for people to use when analyzing their genomic data. A beginning user of their site may feel slightly overwhelmed by the multitudinous options for exploring NCBI's extensive databases, but this website is here to help! It will cover how to decide which BLAST algorithm to use, and then how to use that algorithm.
BLAST stands for Basic Local Alignment Search Tool. To use it, a researcher should submit to the algorithm a sequence of interest. The sequence can be DNA, RNA, or an amino acid chain. The algorithm will then compare the sequence the user submitted with the sequences in its database, and tell the user which database sequence most closely matches the user-submitted sequence. You may have a sequence of interest because you just sequenced a DNA, RNA, or amino acid chain and want to know what organism it comes from or what protein it makes, or you may have a known DNA, RNA, or amino acid sequence from a particular organism and want to know how similar it is to another organism to help determine possible evolutionary relationships.
How to Choose which BLAST Algorithm to Use
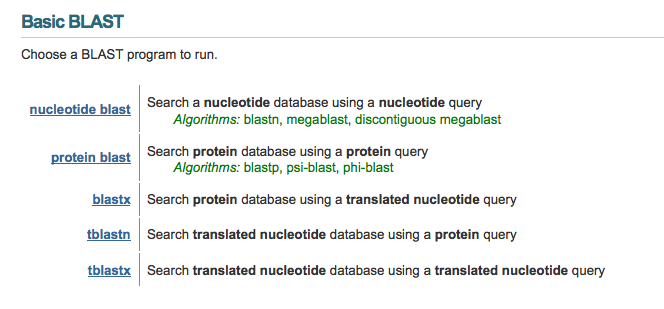
When you go to the BLAST website homepage, you will see the following options:
In this tutorial I will be covering the basics of all of these options, plus the option of a "Blast 2" alignment, for when you have two particular sequences you want to find the similarities and differences between. Here is a quick reference table to help you determine which BLAST option to use:
|
What you have-> |
A DNA or RNA sequence |
An amino acid sequence of a protein |
Two DNA or RNA sequences |
|
Similarities with nucleotide sequence in the database |
N/A |
N/A (or do nucleotide blast two times!) |
|
|
Similarities with proteins in the database |
N/A (or do blastx two times!) |
Similarities with translated nucleotide sequences* in the database |
N/A (or do tblastx two times!) |
|
Similarities with each other |
N/A |
N/A |
bl2seq (also known as BLAST 2) |
*A translated nucleotide sequence means that you submit a DNA or RNA sequence and BLAST will translate it into an amino acid sequence in all six reading frames.
How to do a....
|
|
About BLAST Algorithms
The BLOSUM62 matrix is what all BLAST algorithms use (except the nucleotide BLAST) as a default to align amino acids and determine if two nonidentical amino acids are "positives". This is the BLOSUM62 matrix:
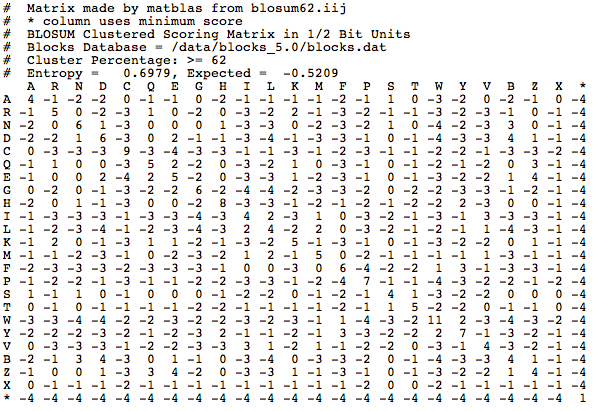
Where did these numbers come from? Each number is referred to as a substitution score. The value of the substitution score is based on amino acid substitutions in real amino acid sequences that are more than 62% identical. The highest substitution scores are generally along the diagonal line that represents where an amino acid matches with itself. That is expected, because typically you would expect that two of the same proteins would have the same amino acid in the same position. Off the diagonal line, positive scores indicate substitutions that occur frequently, and negative scores indicate substitutions that occur infrequently. There are other matrices that you can choose to score your alignments, such as BLOSUM45:
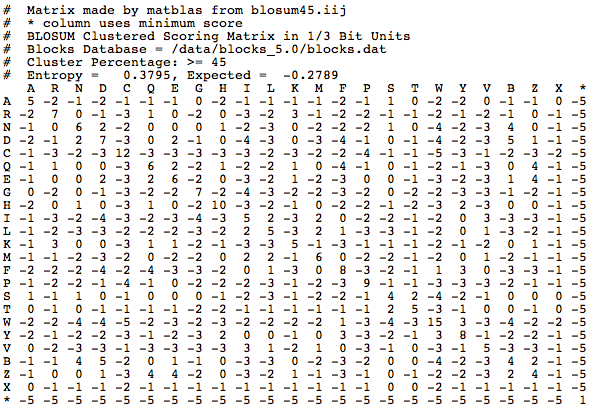
BLAST then finds alignments with high raw scores, which are calculated by adding together the substitution scores of the alignment. The higher the number behind the word "BLOSUM", the more similar you expect your query sequence to be to an exact match in the database. BLOSUM62 is a good place to start.
When using Nucleotide BLAST, the default of Megablast uses a greedy matching algorithm is used to help find optimal matches. Megablast has a gap opening penalty of 0 and is best used for aligning a query sequence that very similar to the expected sequence (like the actual sequenced nucleotide sequence versus the expected sequence). Discontiguous megablast is best for cross-species comparisons. The blastn option has the shortest default word size, and is therefore the most sensitive and will find the most dissimilar matches (you need less exact nucleotide matches in a row to find a blastn match then a megablast match).
Scores and E-Values
Every BLAST result tells you the score and e-value of the alignment. The score is calculated using the raw score (R) with the formula S = [lambda*R - ln(K)] / ln(2). Lambda and K are constants given at the bottom of your BLAST results page, because they change over time. The score measures the similarity between the queried sequence and the found sequence. In protein blasts, the raw score is the total of all the BLOSUM62 substitution scores. In nucleotide blasts, the raw score is the total of all matches, mismatches, and gap penalties. The E-value represents the amount of alignments you would expect to find by chance that have the same score as the alignment you are looking at. The e-value is calculated with the formula E = (query length) * (length of database) * 2^-(S). A good, biologically significant e-value would be 0.05 or less.
Resources
Campbell, A. Malcolm, and Laurie J. Heyer. 2007. Discovering Genomics, Proteomics, and Bioinformatics. Boston: Pearson and Benjamin Cummings.
NCBI. 2000. Substitution Matrices. <
http://www.ncbi.nlm.nih.gov/Education/BLASTinfo/Scoring2.html>. Accessed 5 Oct. 2008.
NCBI. 1997. NCBI BLAST Advanced Options.
<
http://www.ncbi.nlm.nih.gov/blast/full_options.html#blastn>. Accessed 5 Oct. 2008.
Genomics Page
Biology Home Page
Samantha's Home Page
Halorhabdus utahensis Genome Wiki