Coat Variation in the Domestic Dog Is Governed by Variants in Three Genes
This webpage contains a brief review of a paper entitled “Coat Variation in Domestic Dog Is Governed by Variants in Three Genes”. All figures and facts come from this paper by Cadieu et. al 2009. The first section includes a basic summary of the paper. This is followed by a brief summary of each figure and my final conclusions.
Review
Before reviewing the paper, there are a few specific words that should be defined. “Pelage” refers to the dog’s coat and the various characteristics associated with that coat. “Furnishings” refer to the naturally growing longer hair around the ears, the eyes, and the legs in certain breeds (often wire-haired dogs).
In this paper, the authors claim to identify three genes that determine nearly all coat types in domestic dog (~95% of the dogs sampled). The properties of these various coat types include texture (smooth or wiry), length (long, medium, or short), and whether the hair is curly or straight. In order to identify genes linked to these different pelages, the authors choose to analyze breeds where pelage divisions could be made. One data set contained dachshunds with three coat varieties: wire-haired with finishings, smooth, and long-haired without finishings. Another data set used was Portuguese water dogs with curly or wavy hair. The last set was a previously created, diverse dog data set called CanMap that included many dogs (903 dogs) from multiple breeds (80 breeds).
The basic method employed by researchers can be summarized as follows.
- Perform a genome wide association study (GWAS) using SNP microarray data to identify potential genes loci linked a coat characteristic (texture, length, or curly/straight).
- Through statistical analysis of the GWAS data and pedigree data, identify a specific locus for further investigation.
- Further narrow the locus for investigation through fine-mapping.
- Sequence the identified locus and compare the sequences across dog coat phenotypes (hopefully to identify an mutation that correlates with the phenotype).
Through this method, mutations in three genes - RSPO2, FGF5, and KRT71 - were identified. The dachshunds data was used to identify RSPO2, a gene linked to wiry or smooth hair. This dachshunds data was also used to identify FGF5, a gene linked to hair length. Finally, the Portuguese water dog data was used to identify KRT71, a gene linked to curly or straight hair. The CanMap data set was used throughout the investigation as an additional control (or baseline reference for gene expression) and as information used to verify predictions based upon genes.
Through the methods outlined above, the researchers identified RSPO2 as a potential wire-hair gene. The researchers sequenced this gene in seven different breeds and identified at 167bp insertion in the 3’ untranslated region (UTR) in dogs with wire-hair and furnishings. In their set of 704 dogs, 297 of 298 dogs with furnishings had this gene and the 406 dogs without furnishings lacked this gene. Furthermore, previous studies have linked RSPO2 it to pathways involving hair follicles, and a similar mutation in the human hair follicle pathway has been shown to be linked to the coarse hair. To support their claim that this insertion in the UTR could alter RSPO2 expression, the researchers tested the RSPO2 transcript content of the skin on the muzzles of dogs with and without furnishings. They found dogs with furnishings had a 3 fold increase in RSPO2 transcripts.
Previous studies had linked FGF5 to hair length. The data analysis in this paper supported the same conclusion. Through fine-mapping FGF5 was identified as a potential hair length gene. As in previous papers, sequence analysis showed an amino acid change from Cys(95) to Phe to be strongly associated with long-haired dogs. 91% of the long haired dogs were shown to have this allele. Only 3.9% of the short-haired dogs had this allele, and ~30% of medium-haired dogs had this allele.
These same techniques identified KRT71 as a potential curly hair gene. Sequencing identified an Arg(151) to Trp alteration. The authors also cite that alterations in this gene have been linked to curly hair in mice.
Figure 1
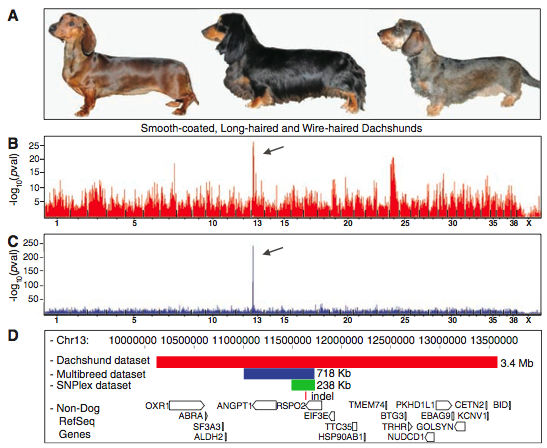
Figure 1 is a visual of the steps used to identify RSPO2 as a wire-haired gene. Presumably the researchers performed these steps for the two other identified genes.
- 1A shows the three different types of dachshunds: smooth-coated, long-haired, and wire-haired.
- 1B shows the microarray data using smooth and long-haired dogs as the controls (and wire-haired as the experimental). The red lines show gene expression that was greater in the experimental than the control. The position on the x axis correlates to that gene’s position within the genome. The arrow points to the greatest p value - which would be the gene with the best association to the experimental phenotype. It is interesting the authors fail to address the group of genes with significant expression levels right before the 25 tick on the x axis.
- 1C shows the results of GWAS using the CanMap data set, divided into control and experimental groups based on the phenotype of wired hair with furnishings. Again, the arrow points to the greatest p value. This data set was used to further verify the results from 1B and rule out false positives due to specific breeds. Note that with this data set, we see a higher resolution in regards to gene positioning along the genome (expression spikes over a smaller locus), and the difference in gene expression is much more pronounced (larger log scale on y axis). These differences could be due to the fact that this was a large and diverse data set (903 dogs composed of 80 breeds).
- 1D shows a mapping of the isolated locus. The red rectangle is the locus associated with wired-hair and furnishings in dachshunds. The blue rectangle shows the locus identified through CanMap. Finally, the green rectangle shows the locus identified through fine-mapping. From this figure, we see the only gene located in the identified locus is RSPO2. We can also see the 167bp insertion within the RSPO2 3’UTR shown by the red indel marker.
I liked and disliked the fact that the authors only provide the equivalent of 1D for subsequent figures. I like it because if I had to choose one panel from figure 1 that I wanted to see for every experiment, it would be 1D. I disliked this decision because of the mere fact that it gives the reader less data to draw their own conclusions from. I understand why the authors chose to do this, given the limited article real-estate in Science.
Figure 2

Figure 2 shows both the mapping and identification of the long-haired gene and curly gene.
- 2A shows the equivalent of figure 1D for the long-haired gene, FGF5. Again, the red rectangle is the locus associated with long hair in dachshunds. The blue rectangle shows the locus identified through CanMap. Finally, the green rectangle shows the locus identified through fine-mapping.The red bar shows the SNP that correlated with hair length differences most often within this gene. It is interesting that an allele for this gene can be isolated to a single SNP.
- 2B shows the different coat types of Portuguese water dogs. Note that the differences are not a clear curly and straight; rather, the phenotype ranges from curly to wavy.
- 2C shows the equivalent of figure 1D for the wavy-haired gene, KRT71. The green rectangle is the locus identified through fine mapping. The red bar with “Best SNP form SNPlex” written to the side is the SNP that most closely correlated with hair length differences in in the fine mapping (based on both the Portuguese water dogs data and the CanMap data). The red bar with “Best SNP form Sequencing” written to the side is the most associated SNP to curly hair identified through sequencing. It is unclear to me why a bar for the locus isolated from the Portuguese water dogs set and from the CanMap data set is absent. From the article it sounds as though they were able to immediately identify a very specific locus which may be what the “Best SNP form SNPlex” bar is trying to show.
Figure 3
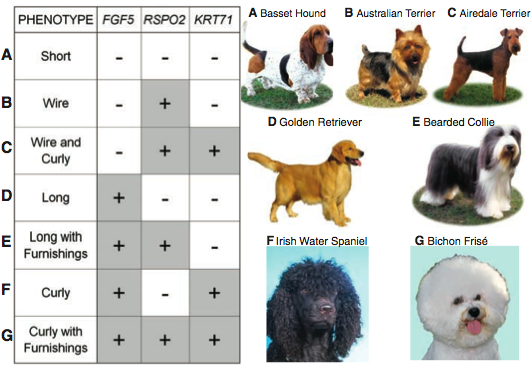
Figure 3 summarizes the whole paper. This is the “splash” figure. It shows how the different combinations of three different genes - at different loci - influence coat phenotype. A “-“ indicates this gene is not present, and a “+” indicates a gene is present. The letters going down the left hand side correlate to the dogs on the right hand side. So, for row A, short hair, an example and visual of a short haired dog is the basset hound on the right side. This figure is very nice because it provides the relevant scientific information along side a visual example. From this figure you know the exact phenotype and genes associated to it.
Conclusions
In their conclusion, the authors state that these three specific variations (mutations) in these three genes explain 95% of the pelage phenotype in the dogs sampled. To me this was astonishing. Three specific mutations on three separate genes could interact to form so many diverse and distinct phenotypes! The authors also argue the short haired domestic dogs (3A) carry the ancestral alleles, as none of the subsequent mutations were identified in wolves. This statement seems to be likely, but I am skeptical because only three wolves were sampled. Finally the authors conclude by stating that most modern breeds originated within the past 200 years. This is remarkably short amount of time to see so much diversity evolve and shows how artificial selection for genes and domestication can usher rapid evolution.
I really enjoyed this paper. It has great figures - you can almost read the whole paper just through the figures and figure legends. There were times I would have liked to see more data - like how often was the KRT71 mutation in the curly haired dogs from the CanMap data set. However overall, the data was convincing - especially with the large CanMap data set and the allele frequency this allowed the authors to compute. The best part of this paper is figure 3 and the conclusions based from it. It is amazing that such diversity can be linked to roughly 169 nucleotides (167+1+1) across three genes!
Works Cited
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, VonHoldt BM, Rhue A, Boyko A, Byers A, Wong A, Mosher DS, Elkahloun AG, Spady TC, André C, Lark KG, Cargill M, Bustamante CD, Wayne RK, Ostrande EA. Coat Variation in the Domestic Dog Is Governed by Variants in Three Genes. Science. 2009 Oct 2;326: 150–153.