Innate immunity is a part of the immune response in which "a variety of innate resistance mechanisms recognize and respond to the presence of a pathogen. It is present in all individuals at all times, does not increase with repeated exposure to a given pathogen, and discriminates between groups of similar pathogens" (Immunobiology, 6/e; Garland Science, 2005). The innate immune system is used by the body to detect and recognize a large number of pathogens and then to discriminate then as nonself. This becomes difficult when the pathogens mutate which can alter the expression and recognition of the pathogen. Host cells however, have developed different innate immune receptors, which can detect different microbial structures.
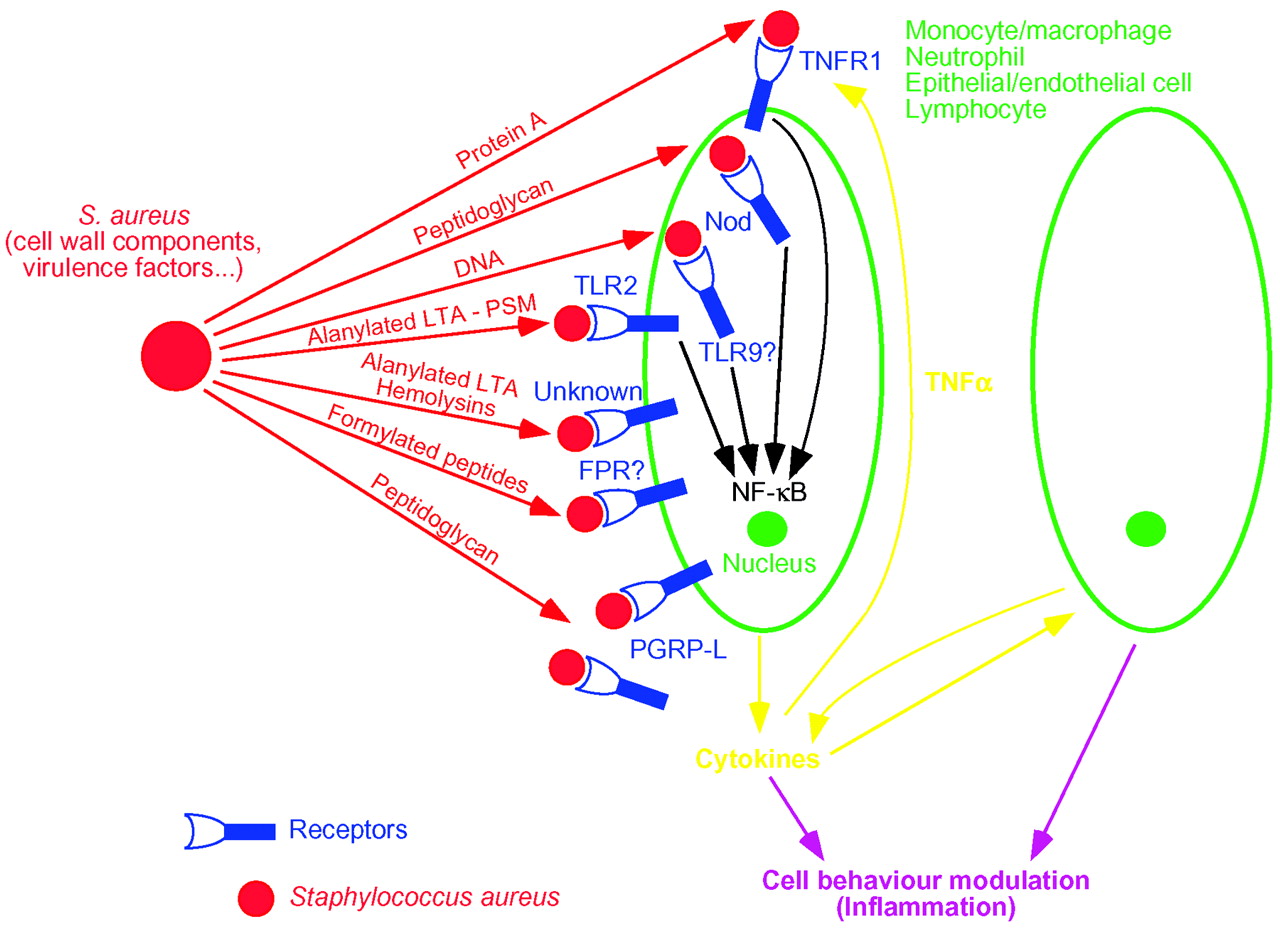
The bacterium, Staphylococcus aureus is a major pathogen that is responsible for a variety of diseases including minor skin infections and life-threatening conditions such as sepsis. As for the host, many host factors have been implicated in the innate detection of staphylococcal components through TLR-2 (Toll-like receptor 2), Nod2 (nucleotide-binding oligomerization domain 2), etc. The staphylococcal virulence factor, protein A, was recently shown to interact directly with TNFR-1 in airway epithelial cells and to reproduce the effects of TNF-alpha. Also, peptidoglycan recognition protein L is an amidase and it inactivates the pro-inflammatory activities of peptidoglycan. Therefore, several innate immunity receptors may be used in host defense against the S. aureus bacterium (including the MRSA strain).
The frequency of gram-positive sepsis has been increasing recently, and this has been associated with the ability of methicillin-resistant strains of S. aureus to colonize intravascular catheters or surgically implanted materials. The outer membrane of staphylococci is made up of peptidoglycan, lipoteichoic acids, and several other toxic components which help to induce septic shock.
Phagocytosis is one of the major mechanisms for combatting the staphylococcal infection. Antibodies neutralize toxins and promote opsonization, but the bacterial capsule and protein A may interfere with phagocytosis.Staphylococci may be hard to kill after phagocytosed because they produce carotenoids and catalase which creates problems for the phagocytic killing mechanisms within the phagolysosome.
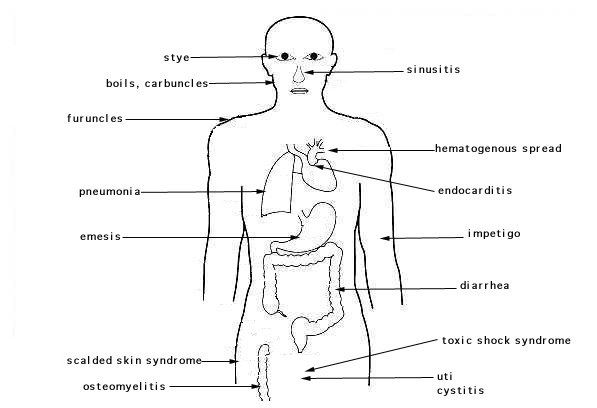
Humans tend to come into contact with the Staphylococcus aureus bacterium by way of external epithelia, and usually through a site of a wound or abrasion. The epithelial surface in one of the initial barrier sites of innate immunity, but the S. aureus bacterium is often able to get past this barrier. Most microorganisms take succeed in breaking through the epithelial surfce are removed by innate immune mechanisms, such as those mentioned in the above paragraph, that function in the underlying tissues. If a large amount of S. aureus pathogen is able to establish a local site of infection and replicate to further transmit the infection throughout the body, an inflammatory response will be started by a number of cell wall-associated and secreted proteins and and cell wall components (such as protein A, hemolysins, peptidoglycan and phenol-soluble modulin). After the inflammatory response begins, many of the cells associated with it, such as macrophages, neutrophils, and dendritic cells, express TLR-2.
The complement system, which is made up of many distinct plasma proteins that react with one another to opsonize pathogens and induce a series of inflammatory responses that help fight infection, is also associate with the innate immune response to MRSA and Staph. aureus. However, the Staphylococcus aureus is able to inactivate the complement cascade by way of the extracellular fibrinogen-binding protein (Efb-C), which is generated by S. aureus strains, and works to block the complement pathway by binding to the thioester-containing domain of the complement C3b protein (Nat. Immunology. 10.1038/ni1450; 2007).
http://cmr.asm.org/content/vol18/issue3/images/large/zcm0030521430003.jpeg