
image: iStockPhoto
This web page was produced as an assignment for an undergraduate
course at Davidson College.
image: iStockPhoto
D. Benjamin Riffe's Genomics Web Page
Home Schizophrenia Paper Review #1 Paper Review #2
Coat Variation in the Domestic Dog Is Governed by Variants in Three Genes
Cadieu et al, 2009
Within the context of domestic dog fur, much is known about the genetic basis of fir color, but very little about length, curl and growth patterns, i.e. “furnishings.” This study took 1000 dogs from 80 breeds as determined by the American Kennel Club (AKC) and observed genome-wide associations among different breeds of dogs, looking specifically at 3 genes: RSPO2, FGF5, AND KRT71. The results of the association studies show the phenotypic diversity that variations in these 3 genes create most coat diversity in purebred dogs.
The paper presents the overall evolutionary history of the modern domestic dog as being one begun 15,000 years ago and highly influenced by selective breeding as well as natural selection. In order to determine marker-based associations within a single breed, observing simple variation within in a breed and using those association regions to look for large variations across breeds.
Using a canine SNP chip, 96 dachshund with wire-haired with furnishings, smooth, and long haired without furnishings, 76 Portuguese water dogs of the curled and wavy haired varieties, and data from 903 dogs from 80 breeds (CanMap) were subjected to the same tests. A genome-wide association study (GWAS) was used within-breed to determine highly associated loci. Furnishings in dachshunds were determined using by using longhaired dogs as controls and known pedigree, with a single-marker GWAS, finding a 718-kb homozyguous haplotype in all breeds within a 3.4 Mb segment on chromosome 13. Fine-mapping found a 238 kb region, the R-spondin-2 (RSPO2) gene, which synergizes with Wnt and activates β-catenin. These genes are associated with hair-follicle growth, hair-follicle tumors, and a change in the pathways is associated with human East-Asian hair type. Dogs with furnishings were found to be homozygous or heterozygous for a 167 bp insert in RSPO2, showing a dominant inheritance pattern. Homozygosity for the ancestral trait was found in all 406 dogs without furnishings. The position of the mutation at the 3‘UTR, which controls mRNA stability, lead a tripling in gene expression in the derived gene.
To find the gene for hair length, identical mapping was used, drawing on previous studies looking at mutations 32 in FGF5 in Welsh corgis causing a fluffy or long-haired phenotype. The results were replicated by looking at the dachshund and CanMap data in CFA32, fine-mapping to find a 67-kb segment homozygous region. This mutation changes a Cys to a Phe. Longhaired dogs contain the TT genotype, and GT and GG genotypes are found in short or wire-haired dogs, showing a recessive inheritance pattern.
The curly coat gene was found using a GWAS and PWDs, finding a SNP association on CFA27 and verified on the CanMap. Fine-mapping found more precisely 2 keratin genes, which had a shared homozygous haplotype. CC codes for non-curly dog fur, TT for curly fur, and in some breeds, all three genotypes can be present. This SNP mutation in KRT71 causes an Arg to Trp mutation, which is non-synonymous. The paper then elaborates on the validity of mutations of keratins as cause for hair growth citing research on mice and ensembl folding results. Sequence changes could cause changes in post-translational processing, which could be the source of the curly hair phenotype.
The paper then shows that combinations of these 3 mutations can account for the fur variation in 95% of sampled dogs. The authors then show the possible combinations and example breeds.
The authors end the paper by placing these mutations in the context of the evolutionary history of the Dog and gray wolf. These 3 mutations are absent in shorthaired dogs and gray wolves, showing that the variants are derived, and transferred between dog breeds by hybridization. Since most modern dog breeds evolved in the last 200 years, this shows a few genotypic variations can create massive phenotypic changes in a short time, especially in cases of artificial selection. Finally, the authors believe this framework will provide insights into mammalian evolution.
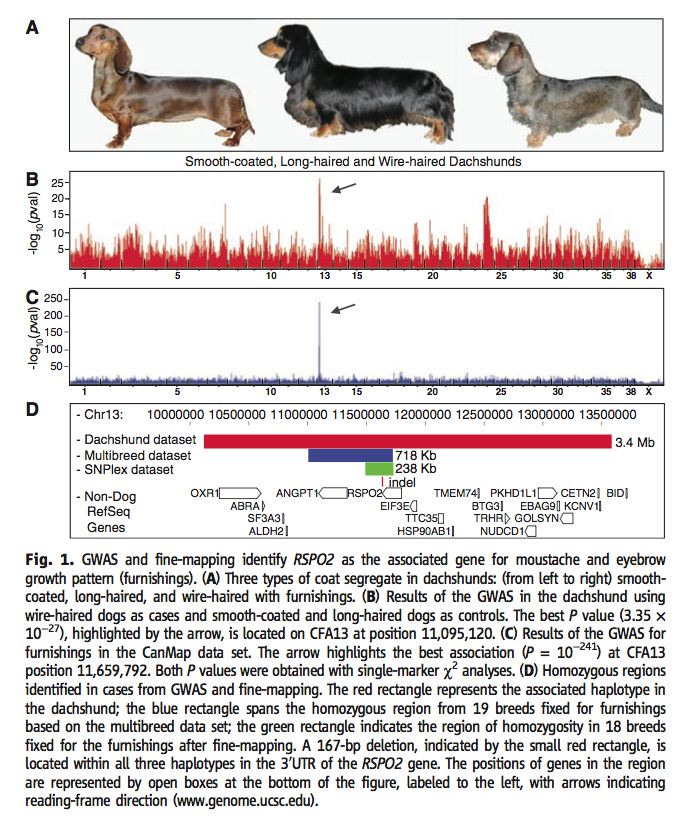
Figure 1† generally looks at the gene responsible for furnishings, RSPO2. Section A shows the 3 phenotypic possibilities for dachshund fur determined by genetic variation of RSPO2. The leftmost dog is the smooth-coated variety, the longhaired dog is found in the middle and the wire-haired dog is rightmost. Section B shows the results of single-marker analysis of GWAS data set and concurrent linkage analysis using the smooth-coated and longhaired as controls. The x-axis displays chromosome number and the y-axis shows the lod score. The high P value of 3.35 x 10-27 at locus 11,095,120 on chromosome 13 is emphasized by the arrow. Section C displays the same GWAS for the furnishings, this time instead using the CanMap data set. The x-axis still shows chromosomes and the y-axis contains the P values, although the scale is much larger. The arrow emphasizes the area of best association with the best P value (10-241) again on chromosome 13 at position 11,659,792. A single marker χ2 analysis was performed to obtain the P values in section B and C. Section D displays position 10,000,000 through 13,500,000 on chromosome 13. The top red bar is the haplotype conserved in the dachshund over 3.4 Mb. The blue bar, or multibreed dataset includes 19 dog breeds and shows their homozygous region for the furnishing phenotype. The lowest and green bar includes data from 18 breeds and shows the post fine-mapping homozygous region. The small red rectangle below indicates a 167-bp deletion found in these three haplotypes at the 3‘UTR of the RSPO2 gene. All genes in the region are shown at the bottom of figure 1, labeled Non-Dog RefSeq Genes.

Figure 2† displays data relating to the other 2 genes studied in this paper: the genes for fur length and curl. Section A is a similar chart to section D of figure 1. The region of chromosome 32 spanning from 7,450,000 to 7,950,000 contains a top red bar containing data from the dachshund data set, showing a 520 Kb section of associated haplotype from 29 long haired dachshunds. The blue bar, or Multibreed dataset, has a length of 125 Kb and is a region found 319 dogs from 31 longhaired breeds. This region is homozygous between all 319 individuals. The lowest, green bar from the SNPlex dataset shows the results of the fine-mapping, which consists of a 67 Kb homozygous region from 39 species. The small red rectangle is the best associated SNP between all datasets found in exon 1 of the FGF5, or fibroblast growth factor 5, gene. Genes found in this region are also found on the lower part of this section in Non-Dog RefSeq Genes, and their reading-frame directions are shown by the arrows.
Section B shows two photos of a curly-haired Portuguese water dog on the left and two photos of a wavy-haired Portuguese water dog. Both sets of photos have a close up and body shot of the coat phenotypes.
Section C looks at the curly gene locus and shows a section of chromosome 27 spanning from position 5,525,000 to 5,555,000. This haplotype analysis shows a 32-Kb homozygous haplotype displayed after the final fine-mapping of 65 individual dogs from 5 curly breeds. The 32-Kb segment is shown similar the previous 2 similar sections as a green bar. The best associated SNP from the SNPlex was found in the homozygous haplotype in exon 2 of the KRT71 gene, and is displayed. Interestingly, a different best SNP was found from sequencing, although both were found in the KRT71 gene. Ensembl gene predictions in the region are displayed below, with reading-frame direction indicated by arrows.

Figure 3† charts a matrix of the three different alleles, FGF5, RSPO2, and KRT71, showing the possible combinations and observed phenotype of those particular combinations. Plus signs designate the non-ancestral genotype and minus signs designate the ancestral. Photos of dog breed with these genotypes and observed phenotypes are pictured to the right of the figure. A basset hound was shown for the typical phenotype short (A), which had the ancestral genes in all three loci. For the wire phenotype (B), which has a the variant RSPO2, an Australian terrier is depicted. For the wire and curly phenotype (C), which has the variants alleles in the RSPO2 and KRT71 genes, an Airedale terrier is pictured. The long phenotype (D) has a the variant FGF5 gene and can be illustrated by the golden retriever. The beareded collie has the variant genes of FGF5 and RSP02 and has the phenotype long with furnishings (E). Finally, the combination of FGF5 and KRT71 variant genes produces the curly phenotype (F), exemplified by the Irish water spaniel, while all three variant genes produce the curly with furnishings phenotype (G), pictured by the bichon Frisé. The variations in phenotype caused by the combinations of these three variant alleles can account for 95% of fur variation in all dogs samples.
Conclusions about the Paper
The authors cleverly used both the extreme variation between particular dog breeds and the similarities within individual breeds to find these three coat variation genes. GWAS can show areas of high association within an individual breeds genome, so that the potential regions for variation become clearer. Coat phenotypes can vary within an individual breed, such as the dachshund, and so finding the regions of variation within a breed becomes simpler. Once the correct locus of variation associated with a particular coat trait becomes evident, additional GWAS with multiple dog breeds can take place to find areas for homozygous haplotypes. Finally fine tuning using the SNPlex dataset focuses down on an even smaller region of the homozygous region until either a SNP or indel is found that can account for the variation.
I appreciate the logic of the approaching to finding variant genes. Using more closely related individuals (within breed) to initially find potential regions of variation and then using multiple breeds and larger datasets to find precise regions of variation is an effective way of honing in on correct variation while being able to ignore all other variation that would be observed when using the multibreed or SNPlex datasets.
The authors present the function of the three varying genes, RSPO2, FGF5, and KRT71, and offer an explanation as to how the ancestral gene functions and how a mutation within the gene could change the coat phenotype as it does. This is an important connection to make between form and function, and while we have the patterns of association, explicitly identifying the function supports their conclusions.
The authors’ conclusions placed these variations in the context of modern dogs, showing a practical application of their research, as well as supporting finding. The photos of the dog breeds, the coats of which had different phenotypes from combinations of variants and ancestral genes (fig. 3) put a face to this research. This research also impresses how fast phenotypic variation can occur with just 200 years, even when looking at genes that in combination can cause huge phenotypic change and evolution.
†All figures from Cadieu et al, 2009.
Works Cited
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, VonHoldt BM, Rhue A, Boyko A, Byers A, et al. Coat Variation in the Domestic Dog Is Governed by Variants in Three Genes. Science. 2009;326:150-153.
Genomics Page
Biology Home Page
beriffe@davidson.edu
Email Questions or Comments.
© Copyright 2011 Department of Biology, Davidson College, Davidson, NC 28035