 photo
photo
compliments of
Dr. Shin Sugiyama
photo
compliments of
Dr. Shin Sugiyama
 Studies at this time also showed that although epibatidine did elicit the
typical response characteristic of an opiod, it was, in fact, a potent
analgesic with a non-opiod mechanism of action. (Daly et al, 1992)
Epibatidine was the first member of this new class of alkaloids and produced
an effect 200 times greater than that of morphine. It was concluded
that a non-opiod mechanism of action was present in epibatidine because
naloxone, an opiod antagonist, did not reduce its effect. It was
also determined that the epibatidine had a negligible affinity for opiod
receptors, again supporting the conclusion that a non-opiod mechanism of
action was at work. (Daly, 1992)
Studies at this time also showed that although epibatidine did elicit the
typical response characteristic of an opiod, it was, in fact, a potent
analgesic with a non-opiod mechanism of action. (Daly et al, 1992)
Epibatidine was the first member of this new class of alkaloids and produced
an effect 200 times greater than that of morphine. It was concluded
that a non-opiod mechanism of action was present in epibatidine because
naloxone, an opiod antagonist, did not reduce its effect. It was
also determined that the epibatidine had a negligible affinity for opiod
receptors, again supporting the conclusion that a non-opiod mechanism of
action was at work. (Daly, 1992)
http://bio.davidson.edu/courses/anphys/2000/Todd/toxintype.htm
After Daly and his associates
published their findings in 1992, a host of other studies were conducted
to determine the mechanism of action followed by this novel alkaloid.
All of these studies reached the conclusion that epibatidine acts as a
nonspecific nicotinic acetylcholine receptor agonist (Ellis, 1999).
What does this mean???
This means that epibatidine binds readily, with an extremely high affinity, to nicotinic acetylcholine receptors present on both central nervous system and ganglionic nerve cells (Qian,1993). To understand why this is important, it is necessary to discuss the basic function of neuronal nicotinic acetylcholine receptors in the body as well as the role that these receptors play in analgesia.
Basic neuronal function occurs through a standard pathway (see image below). First, a nerve impulse travels down the presynaptic cell until it reaches the synaptic terminal. Upon reaching the presynaptic terminal the action potential depolarizes the presynaptic terminal triggering the release of calcium ions into the area. The calcium influx causes the synaptic vesicles, filled with neurotransmitters, to fuse with the presynaptic membrane and release the neurotransmitters into the synaptic cleft that separates the presynaptic and postsynaptic cells. The neurotransmitters then travel across the cleft and fuse with their designated receptor sites. Binding to the receptors, which are embedded in the postsynaptic membrane, causes ion channels to open. The movement of the ions across the membrane will either be excitatory - depolarizing the membrane and leading to propagation of an action potential - or inhibitory - where the membrane becomes hyperpolarized, preventing the propagation of an action potential. The substances bound to the receptors are quickly broken down and the membrane returns to its normal, resting potential. Once the membrane has returned to its normal potential, neurotransmitter can again bind to the receptors and the process is repeated, continuing the propagation of the action potential. (Campbell, 1996)
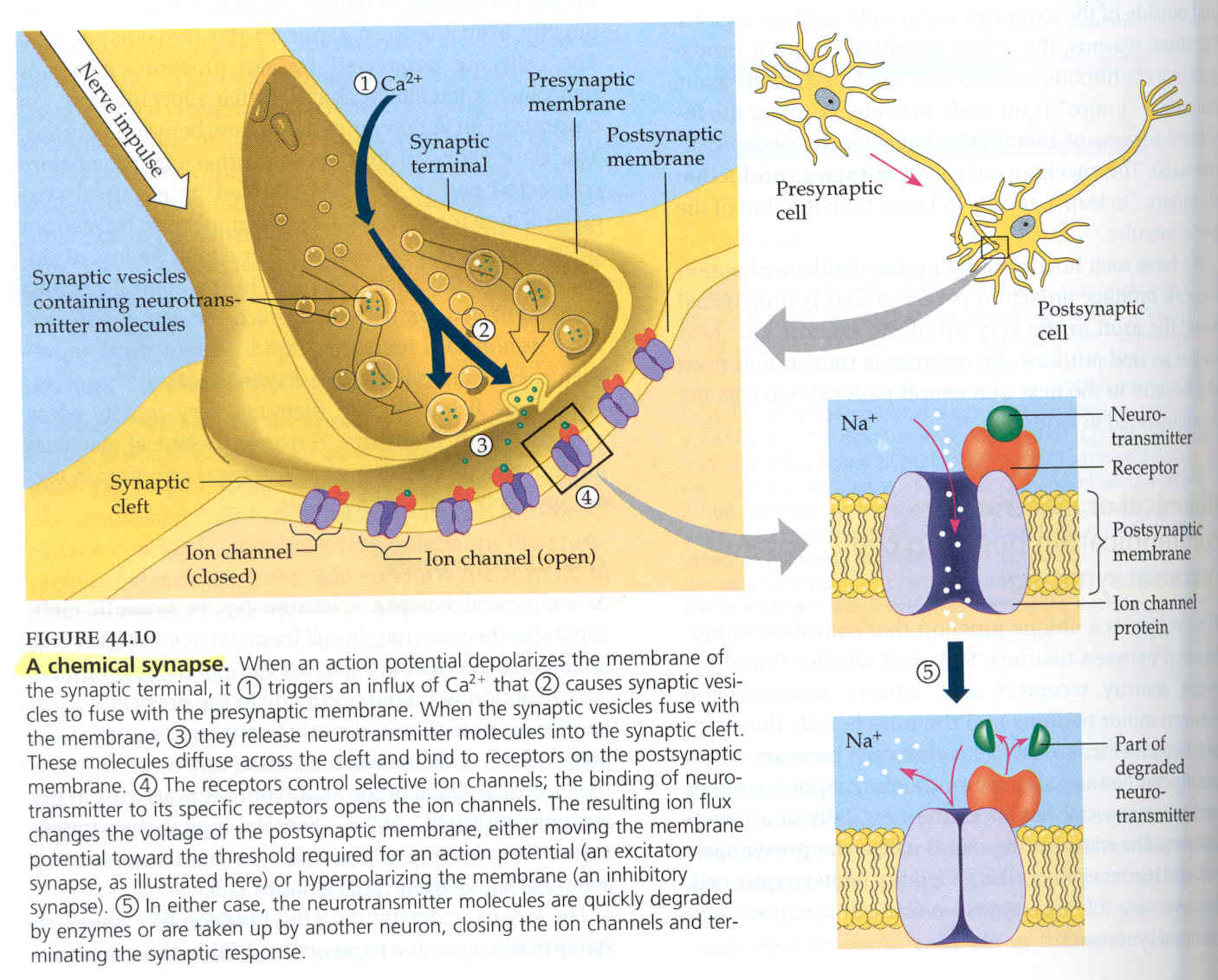
Diagram showing basic process by which nerve impulses are transmitted
throughout the body.
Pictured are the synaptic terminal and cleft (left), presynaptic and
post synaptic neural cells (upper right), and a neurotransmitter receptor
embedded in the postsynaptic membrane (lower right).
(Campbell, 1996)
There are two types of acetylcholine receptors in the body, nicotinic and muscarinic, appropriately named for the compounds that bind to them. Both nicotine and muscarine, a poison derived from mushrooms, mimic the activity of acetylcholine in the body and each stimulates the response of a different acetylcholine receptor. Muscarinic acetylcholine receptors are inhibitory in nature. When muscarine binds to these receptors, hyperpolarization of the membrane occurs, inhibiting the propagation of an action potential. Conversely, the binding of nicotine to nicotinic acetylcholine receptors creates an excitatory response causing rapid depolarization of the membrane and stimulation of the nerve cell. (Lodish et al., 2001)
Analgesia
is defined as the the state of loss of pain sensibility (Adelman, 1999).
The primary site at which analgesics act is the neuromuscular junction.
Nerve transmission can be blocked in two ways: by competitive (non-depolarizing)
or non-competitive (depolarizing) agents (Adelman, 1999). While low levels
of nicotine excite nicotinic acetylcholine receptors, high levels of nicotine
have a reverse effect, inhibiting nerve transmission due to a constant
depolarization of the membrane (Fisher, 1994). Because epibatidine
is similar in structure and function to nicotine, but is120 times more
potent (Qian, 1993), it has an extremely high affinity for nicotinic receptors.
Thus, since epibatidine binds more readily to the nicotinic acetylcholine
receptors than nicotine does, epibatidine will display the same cellular
effects as nicotine at much lower concentrations. Epibatidine, therefore,
acts as an analgesic due to its role in the inhibition of neural transmission
throughout the body.
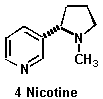
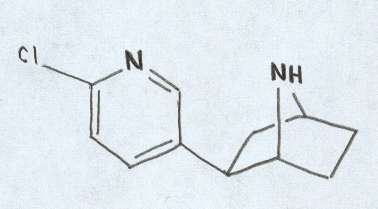
Nicotine
Epibatidine
Nicotine structure obtained from
http://bio.davidson.edu/people/kabernd/seminar/studfold/MUVT/MNVT3.html