Basic Information for the 5 Isocitrate Dehydrogenase Genes
Click on the gene for the following information:
- accession number
- nucleotide sequence and length
- amino acid sequence and length
- molecular weight of protein
- assoicated coenzyme
About the expression vector used-pQE-30 UA
This expression vector was constructed by Qiagen. It contains a viral promoter, a lac operon, an epitope tag, two multiple cloning sites, an ampicillin resistance gene, a start codon, and several stop codons (Qiagen, 2001). The viral promoter allows for mass production of the protein in E. coli (2001). The lac operon allows for regulation of transcription of the insert (2001). The ampicillin gene allows for selection of transformed E. coli. The multiple cloning sites allows for selection of the plasmids with the insert in the correct orientation (2001).
Cloning using the pQE-30 UA vector
The Taq polymerase used in the PCR reaction to amplify the gene that will be inserted adds the nucleotide, A, to the 3' end of all of its products (Qiagen, 2001). In cloning, the PCR product with it's 3' A overhang is added to a prelinearized pQE-30 UA vector that has a 3' U overhang on each end (2001). In the ligation reaction the U overhangs in the plasmids and the A overhangs in the insert bind to each other and then ligase anneals the gaps in the backbone (2001). By using the UA overhangs, the addition of a linker to the primer nor restriction digest of the plasmid needs to be performed (2001).
View the sequence of the expression plasmid and learn more about the plasmid here: pQE-30 UA
What is PCR?
PCR is short for the polymerase chain reaction. It is a molecular biology method used to amplify sequences of DNA using the following steps:(Campbell, 2002)
1. Heat the genomic DNA and the primer to 95 degrees Celsius for 5 minutes to denature the genomic DNA.
2. Lower the temperature to 40 degrees Celsius for 1 minute to let the primers anneal to their corresponding sequence on the genomic DNA.
3. Raise the temperature to 72 degrees Celsius for 1 minute to let Taq polymerase replicate the genomic DNA. Taq polymerases extends the primers at the 3' end.
4. Raise the temperature to 95 degrees Celsius for 1 minute to denature the newly synthesized PCR product.
5. Repeat steps 2-4, 20-30 times. Do not denature the DNA at the end of the final cycle.
6. Isolate the PCR products.
Designing the PCR primers used to to amplify the 5 isocitrate dehydrogenase genes
PCR requires two sequence specific primers, a 5' and 3' primer, to be made for each gene. The 5' primer consists of a sequence that is identical to the 5' end of gene. The 3' primer consists of a sequence that is the reverse complement of the 3' end of the gene. Thus the 5' primer binds to the 3' end of the template strand and the 3' primer binds to the 3' end of the gene (Figure 1). Each primer needs to be between 18-30 base pairs to ensure specificity (Campbell, 2002). Each primer should also anneal to the gene within 44-46 degrees Celsius (2002). If all the primers anneal at around the same temperature, then all five of the PCR reactions can be run at the same time (2002). Since we used a plasmid that has U overhangs at the cloning site, no restriction sites or flanking sequences needed to be added to the ends of the primer (Qiagen, 2001). Thus the 5' primer sequence should begin at the start codon and the 3' primer sequence should begin at the stop codon ( reverse complement).
Figure 1: PCR Primers
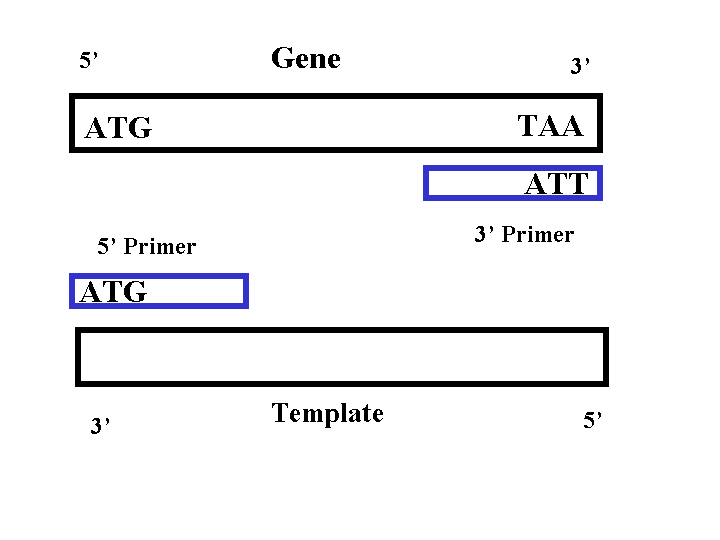
Requirements for primers: (Campbell, 2002)
Melting Temperature Tm (temperature at which the primers anneal): 44-46°C
Length: 18-30
GC content: 40-60%
Primers for PCR of the 5 isocitrate dehydrogenase genes: Final Primers
How to determine the orientation of the insert in the plasmid
Restriction enzyme sites located within the gene and the plasmid will be used to determine the orientation of the insert. The length of the restriction fragments will depend on the orientation of the gene (Figures 2, 3, and 4).
Legend to figures 2 and 3
Figure 2: Plasmid with insert in correct orientation
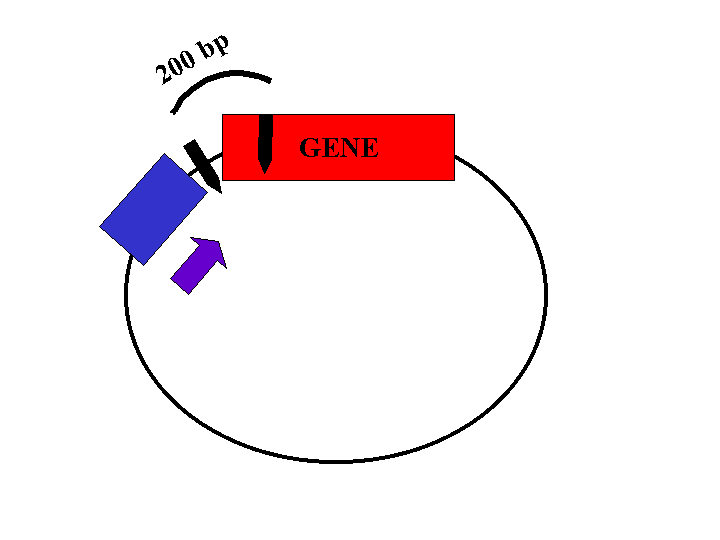
Figure 3: Plasmid with insert in wrong orientation
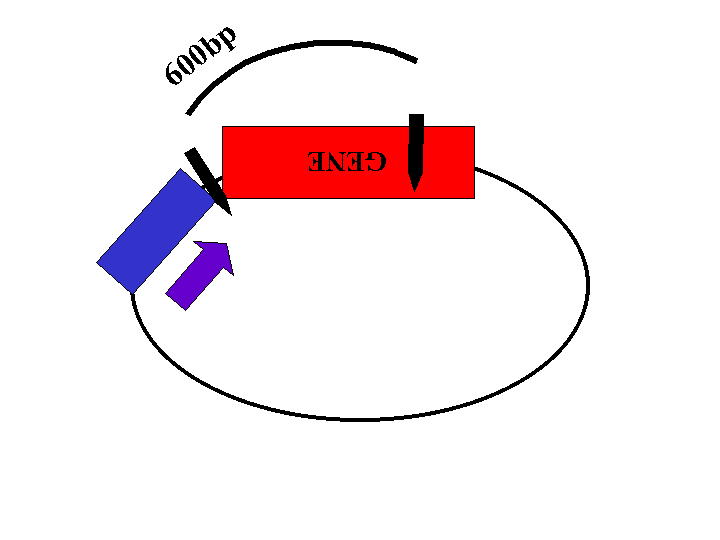
Figure 4: Southern Blot of Plasmid with Insert
When performing restriction enzyme digests it is easier to work with only one enzyme (Campbell, 2002). Every restriction enzyme has an optimal pH and salt concentration (2002). If the reactions are not run under optimal conditions star activity may occur (2002). Star activity is when the enzyme loses its specificity (2002). If two enzymes are used differences in optimal pH and salt concentrations must be considered (2002). Since the restriction fragments will be immediately run on a gel to determine their lengths, base pairing between similar sticky ends was not a factor (2002). The restriction enyzmes used should be found in the multiple cloning site of the plasmid and cut the insert once. The following restriction sites are found int the mutliple cloning site of the pQE-30UA vector: BamHI, PmlI, EcoRV, BglII, SacI, KpnI, SmaI, XmaI, SalI, StuI, PstI, and Hind III (Qiagen, 2001). The following restriction enzymes were at our disposal: BamHI, ClaI, EcoRI, EcoRV, HindIII, KpnI, PstI, and SalI. The restriction enzymes that cut the gene can be determined by looking at a restriction map. Four of the five genes had restriction sites that were also present in one of the multiple cloning sites of the plasmid. Unfortunately none of the restriction sites in the IDP3 gene matched the restriction sites found in the multiple cloning sites of the plasmid. Thus for IDP3 two different restriction enzymes needed to be used. There is only one enzyme, ClaI, that was available to us and also cut the IDP3 gene. It would be nice if the enzyme used to cut the plasmid worked with the same buffer as ClaI or if both enzymes worked well with the multi core buffer. The ClaI buffer does not match any of the other buffers specified for the restriction enzymes at our disposial but both ClaI and HindIII have 100 percent activity with the multicore buffer. Thus ClaI and HindIII will be used to determine the orientation of the IDP3 insert, EcoRV will be used to determine the orientation of the IDH1 insert, and BglII will be used to determine the orientation of the IDH2, IDP1, and IDP2 inserts.
Click on the following link to see restriction maps for the five IDH genes and the fragment sizes created by the five restriction enzymes that will be used in determining the orientation of the plasmid: Restriction maps
Click on the following link to see the expected fragment sizes that result depending on the orientation of the insert: Restriction enzyme and fragment size info
Click on the following link to see how to run a restriction digest: Restriction digests
Since the insert is located several nucleotides down from the transcription site. The isocitrate dehydrogenase proteins that will be transcribed in the plasmid will have several amino acids added to the beginning of the protein. The following link displays the amino acid sequence of the transcription product that results when each of the five genes are inserted into the plasmid: Fusion Proteins
Ending Notes:
This web page specifically describes how the five isocitrate dehydrogenase genes were cloned into an expression vector. The methods described here can be used to clone any gene for which the gene sequence is known.
Dr. C's Molecular Biology Home Page
send comments to lirobinson@davidson.edu